| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
![]()
“hex /heks/ v. & n. US – v. 1. practise witchcraft. 2. bewitch. – n. 1. a magic spell. 2. a witch. [GK hex six].” (The Concise Oxford Dictionary).
“Why, sometimes I've believed as many as six impossible things before breakfast.” (Lewis Carroll).
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex is an interactive molecular graphics program for calculating and displaying feasible docking modes of pairs of protein and DNA molecules. Hex can also calculate protein-ligand docking, assuming the ligand is rigid, and it can superpose pairs of molecules using only knowledge of their 3D shapes. Hex has been available for about 12 years now, but as far as I know, it is still the only docking and superpostion program to use spherical polar Fourier (SPF) correlations to accelerate the calculations, and its still one of the few docking programs which has built-in graphics to view the results. Also, as far as I know, it is the first protein docking program to be able to use modern graphics processor units (GPUs) to accelerate the calculations.
The graphical nature of Hex came about largely because I wanted to visualise the results of such docking calculations in a natural and seamless way, without having to export unmanageably many (and usually quite big) coordinate files to one of the many existing molecular graphics programs. For this reason, the graphical capabilities in Hex are generally relatively primitive compared to professional molecular graphics packages, but the main aim here is to do docking, not to make publication-quality images.
In Hex's docking calculations, each molecule is modelled using 3D expansions of real orthogonal spherical polar basis functions to encode both surface shape and electrostatic charge and potential distributions. Essentially, this allows each property to be represented by a vector of coefficients (which are the components of the basis functions). Hex represents the surface shapes of proteins using a two-term surface skin plus van der Waals steric density model, whereas the electrostatic model is derived from classical electrostatic theory. By writing expressions for the overlap of pairs of parametric functions, one can obtain an overall docking score as a function of the six degrees of freedom in a rigid body docking search. With suitable scaling factors, this docking score can be interpreted as an interaction energy, which we seek to minimise.
Due to the special orthogonality property of the basis functions, the correlation (or overlap as a function of translation/rotation operations) between a pair of 3D functions can be calculated using expressions which involve only the original expansion coefficients. In many respects, this approach is similar to conventional fast Fourier transform (FFT) docking methods which use Cartesian grid representations of protein shape and other properties, and which then use translational FFTs to perform the docking correlations. However, the Cartesian grid approach only accelerates a docking search in three translational degrees of freedom whereas the SPF approach allows the effect of rotations and translations to be calculated directly from the original expansion coefficients.
Even though the FFT part of a docking search may be fast, the overall speed of calculation still depends very much on the initial "set-up" costs and the final "post-processing costs" of filtering and perhaps clustering the results. Hex is fast because it uses FFT correlations as much as possible, and because the "set-up" costs are much lower in the SPF approach than in Cartesian grid-based approaches. It also turns out that the FFT part of the calculation maps very well to the GPU hardware. Thus, further speed-ups can be expected if you have a suitable graphics card.
Although it is not always easy to compare the performance of different docking algorithms because a lot depends on the size of the translational or rotation steps used, for example, I would still claim that Hex is at least 10 times faster than conventional FFT docking algorithms. Hex is also very easy to use. However, to use Hex most effectively, it can sometimes require some thought when setting up the calculation, especially when setting up the starting orientations of the proteins to be docked.
In the spherical polar approach, it is natural to assign the six rigid body degrees of freedom as five Euler rotation angles and an intermolecular separation. Thus, in complete contrast to Cartesian based FFT approaches, the rotational part of a docking search is the “easy bit” and modelling translations becomes the “hard part.” Fortunately, however, only a few translations (typically about 40 steps of 0.75 Ångstrom) are required to complete a six dimensional docking search. One advantage of the spherical polar approach is that it is easy to constrain the docking search to one or both binding sites, when this knowledge is available, simply by constraining one or two of the angular degrees of freedom. This can reduce docking times to a matter of minutes on a modern workstation.
As of Hex version 5.0, the rotational part of the search may be accelerated by one-dimensional (1D), three-dimensional (3D), or five-dimensional (5D) rotational correlations. Earlier versions just used multiple 1D correlations. The 1D correlations are fast, but the 3D correlations in Hex 5.0 are about twice as fast. However, for basic shape-based docking, 3D is also (counter-intuitively) faster than 5D, largely due to the relatively high "set-up" cost of the 5D correlation expression.
As of Hex version 6.0, the 1D and 3D calculations may optionally be performed on one or more GPUs, which can give a considerable speed-up compared to using conventional CPUs. In this case (also counter-intuitively), 1D correlations are much faster than 3D correlations on the GPU. Additionally, the CPU-based calculations have also been re-written to use multi-threading in order to support parallelisation on both Windows and and Linux-based multi-core systems. Thus, significant performance improvements can be expected from version 6.0 if you have suitable hardware.
Closely related to the protein docking problem is the molecular similarity problem - i.e. how to find the relative orientation of a pair of similar molecules such that some measure of the similarity (difference) between the molecules is maximised (minimised). Both problems involve translating and rotating one or both molecules into the desired orientation. However, to a first approximation, the similarity problem can be reduced to a three dimensional rotational search by initially placing both molecules in a common coordinate system. Although Hex will remain primarily a docking program, the 3D superposition calculations implemented in Hex demonstrate the potential for performing fast 3D superpositions using the SPF correlation approach. Work is in progress to develop this approach further as a separate program for high throughput ligand screening.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
This is a bug-fix release from version 6.1.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
This Manual attempts to describe the main features of Hex by
(a) mentioning each feature at least once, and (b) by giving some examples
of how to use the program most effectively.
In the following sections, italic text is used to refer to Menu Item
and Button Names, or other important concepts within the
program. Typewriter text is used to
indicate a sequence of menu selections or button actions
that are required to
perform a particular function. This type of text is also used when listing
the contents of some of the example files provided with the program.
Bold face text is used to highlight
file or directory names that refer to the installation and
use of Hex.
If you really want to read the whole of the Manual, print out the
PDF version.
Otherwise, browse it on-line as HTML using Hex's Help button.
The graphical user interface (GUI) in Hex is intended to be easy to use. Generally, most actions cause an immediate effect on the display so that once you've loaded a protein or DNA molecule (preferably two such molecules), most of the program's features can be understood by experiment. If you're comfortable with this approach, go right ahead. Much of this Manual can probably be skipped. Just keep an eye on the Messages Window for any information messages. The parts that everyone should read are the sections on Docking (see section Docking Molecules), and Superposition (see section Molecular Matching). If you want to try more than just one or two docking calculations, you should also look at the section on using macros (see section Macros) and the contents of the examples directory (see section Examples Directory). It should be noted that algorithmic details of the docking and superposition calculations are not given here. Its assumed that you have copies of the relevant publications (see section References).
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
If you haven't already done so, please download and install Hex from Hex's Home Page. See section Installation Guide for details. It should be easier to follow this Manual if you have Hex up and running in front of you. Been there, done it? Great! Lets get started... Hex reads protein and DNA molecular structures from PDB-format files. As of version 5.0, Hex can also read SDF-format small-molecule structure files. PDB files can be downloaded from the main Protein Data Bank repository at Rutgers University. Up to three input files can be loaded into Hex at any one time. These are treated as a receptor, a ligand and a reference complex. We'll ignore these distinctions for now and just load a single protein. Go to the File menu and select:
File ... Open ... Receptor |
When you release the mouse button, a new File Selection menu panel should appear. Edit the Filter text area to specify the directory containing your PDB file(s) and press the Filter button. Alternatively, you could navigate to the hex/examples directory and load one of the provided example PDB files. You should now see one or more PDB files listed in the Files box (if not, use the File Selection controls to navigate to the hex/examples directory). Pick a PDB file by double clicking on it (or by highlighting it and picking OK). You should now see a skeletal display of the molecule(s) from your chosen PDB file. Figure 1 below shows the scene obtained after loading the example file 3hfl_fv.pdb, which is the Fv fragment of the HyHel-5 antibody.
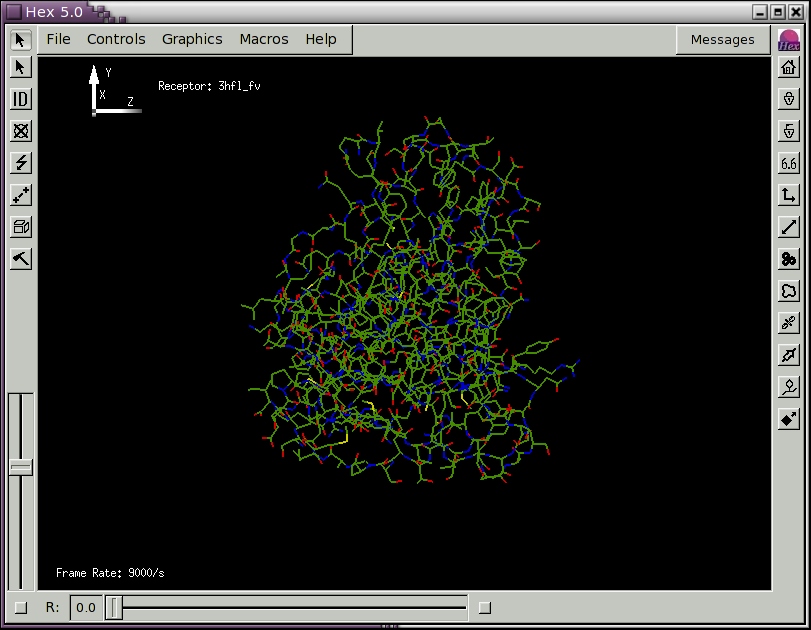
Figure 1. The initial Hex scene showing the HyHel-5 antibody Fv domain as a molecular skeleton.
In Figure 1, you will also see a small
graphic in the top left of the scene that represents the
(x,y,z) coordinate axes.
Note that the z-axis
points towards the right and the x-axis points away from you, into the
scene.
Hex uses this convention because most displays have a greater width
than height and when two molecules are loaded it is convenient to assign
the z-direction to the intermolecular axis. Also, looking at a pair of
molecules “side-on” somehow seems more natural.
Anyway, you can translate and rotate the scene with the mouse buttons.
Assuming you have a three-button mouse, dragging with button
1 (left button) translates the scene, and dragging with Button 2 (middle
button) rotates it. This rotation is always about an axis perpendicular
to the direction of motion of Button 2. The right button also rotates
the scene, but about different axes. A right-left motion of this button
gives an anticlockwise rotation.
You will probably find that Button 2 feels
more natural for most movements but that occasionally Button 3 is needed
to complete a manoeuvre. If you have a one-button mouse (e.g. on a Mac),
you can hold down the Ctrl or Meta (Command/Special) keys with
Button 1 (which might be the only button) to simulate buttons 2 and 3,
respectively.
On Linux, the Ctrl or Alt keys do the same thing.
Some window managers (e.g. Gnome) may interpret Alt-drag as a
window movement shortcut. There should be a window manager option
that allows this key binding to be changed to make the Alt available
to Hex.
The Slider on the lower left
border may be used to zoom the scene in and out. The other borders around
the graphics window contain various pull-down menus and control buttons.
These are described in more detail below.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The column of buttons on the right-hand border of the main window implement handy or frequently used operations. For example, Hex stores a Home Position for the scene. Pressing the House button at the top right border resets the scene to the current home position. If you have oriented the molecule into a view that you like, you can make this orientation the Home Position position by pressing the Lock button, below the Home button. The Unlock button resets the Home Position to its the original setting (z-axis to the right, etc.). The next button (the “6.6” icon) is a text toggle, to control the display of summary text in the graphics window. The Axes button (two arrows at right angles) toggles the display of the coordinate axes (useful for screen shots). Below this is the Intermolecular Axis button (a double-headed arrow). This draws a white line between the centroids of the receptor and ligand molecules. If only one molecule has been loaded, a short white line is still drawn along the z-axis.
The Solid Models button toggles the display of solid models, the
default being van Der Waals spheres. The type of solid model to display
may be selected from the Solid Models control panel
(see section Solid Models).
The Solid Surface button toggles the calculation and display
of solid surfaces (see section Solid Surfaces).
Similarly, the Harmonics button (which looks like a balloon),
toggles the calculation and display of spherical harmonic
molecular surfaces (see section Spherical Harmonic Surfaces),
and the Cartoon button (which is supposed to look like part
of a protein ribbon) toggles the cartoon displays.
The Sidechain button toggles the display of protein sidechains and DNA bases. With large molecules, rotating and translating the scene can be much faster if only the backbone trace is drawn. This is especially true if solid models or surface meshes are being displayed. The final Solid Motion button (the black square with an arrow) can be used to toggle whether solid shapes or just bond skeletons are drawn when moving molecules within the scene.
As you might expect, Hex is normally used to display two molecules at once. Go to the File menu and click on:
File ... Open ... Ligand |
to open another molecule. You could open the same molecule for the ligand as you have opened for the receptor, in which case the two molecules will be superposed. Figure 2, below, shows the result of loading the example lysozyme structure 3hfl_ly.pdb, which is the natural antigen (ligand) of the HyHel-5 antobody. In this example, the R slider bar on the bottom border was used to separate the molecules, and the van der Waals display mode and intermolecular axis were enabled using the right-hand border short-cut buttons. Try experimenting with the other short-cut buttons. You should now find that the mouse buttons translate and rotate both molecules in the scene.
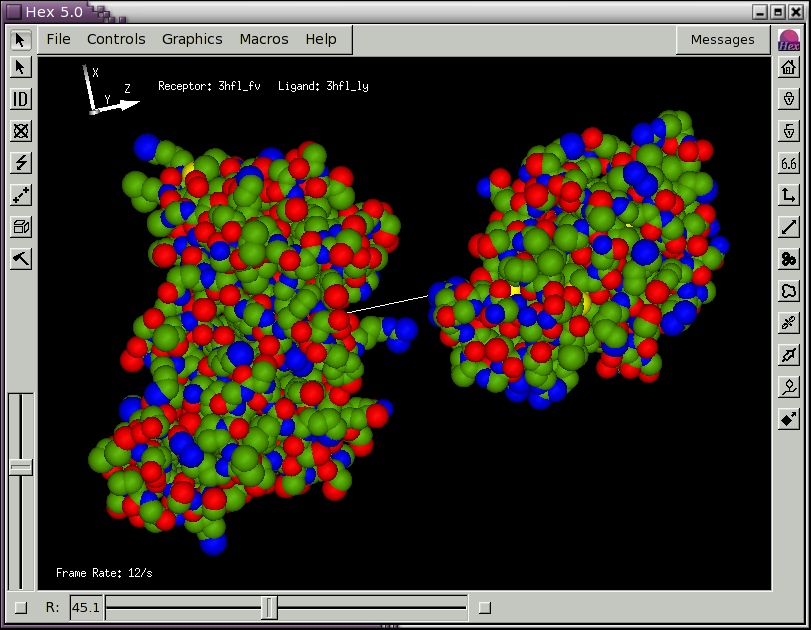
Figure 2. A Hex scene showing the HyHel-5 antibody Fv domain and lysozyme in van der Waals mode, and with the intermolecular axis drawn in white.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The column of buttons on the left hand border of the main window implement various picking and editing functions. These operations differ from the short-cut buttons in that they generally involve changing the operating mode of the mouse. The inset icon in the top left border displays the current mouse mode. The default, Pointer Mode (a left arrow), represents the basic scene manipulation mode, described above. You can revert to this mode by pressing the Left Arrow button, which is the first button in the left-hand border.
The next ID button selects
atom picking mode. In this mode, picking an atom (i.e. pressing and releasing
Button 1 over an atom) will identify the atom by displaying its chain,
residue and atom numbers. The atom coordinates and charge (Q) will also
be displayed in the Messages window.
The Graphics ... Fonts menu can be used to select the font
in which atom labels are displayed.
In stereo displays, picking uses the right-eye
object coordinates, since most people are right-eye-dominant.
Picking the same atom again will remove the atom ID label.
The Hammer button (the last button in this border) will clear all
picked ID's. This button generally acts to undo, or kill, the
previous operation(s), depending on the current mouse mode.
Below the ID button is the Manoeuvring Molecules button
(which looks like a lightning bolt),
and below this is the Origin Editing button (a line with two crosses).
This is followed by the Clipping Mode button (three faces of a cube)
which controls scene clipping (see section Clipping the Scene).
Manoeuvring molecules (also called editing) is something you may need to do before running a docking calculation. For example, before docking, you should always ensure that the molecules are located reasonably close to each other and that their centroids are a reasonable distance (e.g. 30 Ångstroms) apart. The white intermolecular axis connects the default centroids of each molecule. If you have a good idea about the binding epitopes of one or both proteins, then it would be a good idea to rotate one or both proteins such that the intermolecular axis passes through the centre of the epitope. Before you start a docking calculation, you should always consider carefully the locations of the molecular centroids and the relative orientations of your proteins with respect to the intermolecular axis. The Origin Editing button allows you to move these centroids and the Orientation Editing button allows you to rotate each molecule about its centroid and to translate each molecule individually relative to the scene. These operations are described in more detail in See section Manoeuvring Molecules. Note that the R slider bar can be used to change the intermolecular separation permanently, as described above, but it is recommended that you use this control only for temporary changes to the scene (e.g. when viewing the result of docking or superposition calculations) and that you make permanent changes only in editing mode.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
All of the features in Hex are controlled using the GUI components.
However, when you place the mouse in the main graphics window,
several keyboard keys can be used as additional short-cut ways of
controlling the program.
For example, F toggles between full-screen
and windowed mode,
G toggles the GUI border buttons and sliders in full-screen mode,
S toggles stereographics on or off (if stereo is available),
P toggles between perspective
and orthographic projections, and B toggles a checker-board background
(which can help enhance perspective projections).
Hitting the escape key (Esc)
or typing Ctrl/Q will exit the program.
Typing Ctrl/C will interrupt and exit the program even if it is
in the middle of a calculation.
When viewing the calculated orientations from a docking run, the
keypad Page/Down and Page/Up
keys may be used to scroll through the predicted orientations,
and the Home and End keys may be used to jump to the initial
(before docking) and last docked orientations, respectively.
The keyboard arrow keys
are also mapped to these actions
(Down, Up, Left, Right, respectively).
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex can display protein and DNA molecules in several ways. The simplest display styles are controlled by the settings in the Molecule Control panel and some of the short-cut buttons on the right-hand border. Molecules can also be displayed as solid models (see section Solid Models), solid surfaces (see section Solid Surfaces), spherical harmonic surfaces (see section Spherical Harmonic Surfaces), and dot surfaces (see section Dot Surfaces panel). These display types are controlled by additional control panels under the main Graphics menu. This menu also contains options to allow the foreground and background colours to be selected, the position and colour of a directional light to be modified, and a simple Fog effect to be applied.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The basic appearance of protein and DNA molecules is controlled by the Molecule Control panel. The default style is to draw all covalent bonds as a skeleton, with each half-bond colour-coded by atom type. Probably the two most useful controls in this panel are the Show Sidechains toggle (which displays a protein in backbone-only or backbone+sidechain mode) and the Colour Scheme selector, which allows a limited degree of control over the colours used to draw each molecule. Line widths may be changed using the Bond Widths control.
When a protein or DNA molecule is loaded, Hex adds any missing polar hydrogen atoms using a set of templates from the atom_templates.dat file in the data directory. Polar hydrogens can be displayed using the Show Hydrogens toggle. The Show Sidechains toggle has an equivalent short-cut button on the right-hand border of the main window (which is supposed to look like a phenylalanine residue). Setting Show Sidechains to off (which also turns off hydrogen atom display) makes manipulating complex scenes easier.
When viewing large molecules, its sometimes useful to draw only those atoms within a given distance of the molecular origin. The Apply Radial Cutoff button may be used to enable this behaviour, and the Radial Cutoff slider may be used to adjust the radial distance threshold. Distances are relative to the current scene rotation centre, which may be changed using Origin Editing button in the left-hand border (the crossed circle icon).
The Colour Scheme selector allows the molecular skeleton to be coloured from a fixed palette of colours or by using very simple commands in a text file to specify particular colours for specific residues. Some examples of colour files can be found in the examples directory. The general format for a colour file is:
chain first_residue last_residue colour_name |
where colour_name
is any one of the X11 colour names.
The colour names may be found by listing the
rgb_colours.dat file in the data directory.
Alternatively, each time a colour is picked from the Colour Chart
its name is printed to the terminal window.
Shown below is an example colour file 3hfl.col
from the examples
directory, which specifies the colours
to use when drawing an antibody/antigen complex. Subsets of the residues in
each chain are assigned different colours to highlight the positions of the
antibody hypervariable loops:
L 1 214 grey L 25 33 red L 90 97 blue H 1 225 RoyalBlue1 H 25 33 red H 52 56 yellow H 95 102 green Y 1 999 OrangeRed Y 41 53 yellow Y 67 70 red Y 84 84 blue |
Colour files may also be used to colour atoms explicitly, using the syntax:
atom_name first_atom last_atom colour_name |
The Molecule Control panel also contains toggles to display
molecular centroids and average molecular ellipsoids.
The ellipsoids are
calculated from low order (L=2) spherical harmonic surfaces.
This control panel also allows the display of any
crystallographically related molecules whose coordinates are not given
explicitly in the PDB file. Use the Symmetry Type
selector to select these additional symmetry elements.
Selecting Crystal causes the scene to be generated using the
SMTRY transformations from the PDB file,
Biological selects the transformations from the BIOMT records,
and None (the default) just uses the explicit atoms coordinates as
usual.
Note that atom coordinates are only replicated at display time using OpenGL
display lists, so that even quite large molecules can be displayed
reasonably quickly.
A fun one to try is the tobacco mosaic virus (PDB code 1RMV), especially when
low resolution harmonic surfaces are enabled. Another nice structure is the
viral coat protein of the phi-X174 bacteriophage (PDB code 1CD3), which just
happens to be the 10,000th entry in the PDB.
By selecting the File ... Save ... Symmetry ... Biological option,
Hex will write out all the symmetry-related atom
coordinates when saving a structure to disc. This is useful
in cases where the biologically active molecule is a dimer (or has some
sort of symmetry) but only the minimal coordinate set appears in the PDB
data file. Some further editing of the output file (e.g. to rename
duplicated chains) may be required if the chains are to be individually
coloured, for example.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The Graphics ... Solid Models control panel contains several options
for drawing lit solid models of molecular scenes, including van der Waals
spheres, licorice, and tube representations. Aromatic rings may be
drawn as hexagonal or pentagonal “blocks”, which can produce some
interesting displays, especially when viewing DNA molecules.
The position and colour of the light may be controlled using the controls
in the Graphics ... Lighting control panel.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The Graphics ... Cartoons control panel contains several options
for drawing proteins as ribbon cartoons. The default behaviour is to draw
a cartoon over the molecular skeleton. To view only the cartoon, you
can disable the molecular skeleton using the Display Molecule button in
the Controls ... Molecule panel. There are several options on the
Cartoon Control panel which allow the colour, width, and thickness
of ribbon cartoons to be controlled. Different parts of the ribbon can
also be coloured according to secondary structure type (i.e. helix, sheet,
turn). The type of secondary structure is currently calculated using
Frishman and Argos'
Stride program.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Solid molecular surfaces may be drawn using the
Graphics ... Solid Surfaces control panel. These surfaces are
calculated using a novel marching tetragons algorithm to contour
a Gaussian density representation of the atoms of each molecule.
The surface skins used in the docking correlations
are calculated using this Gaussian density approach.
The Colour Mode selector allows surfaces to be
coloured by atom colour (the default), electrostatic
potential or charge density, or by using the “classic” blue-red colour
scheme used in earlier versions of Hex.
For electrostatic surfaces, a colour ramp is calculated
which initialy shows positive potentials/charges in blue, negative
values in red, and intermediate values in white. The
Surfaces ... Colour Ramp button may be used to activate a
simple graphical controller for the colour ramp.
The electrostatic potential and charge density displays are calculated
from the in vacuo global charge density expansion using the
current speherical polar docking expansion order, N.
The colours in a charge density display may appear somewhat “washed-out” compared to (perhaps more familiar) potential displays. This is because the potential is calculated directly from the charge density using Poisson's equation, and the del-squared form of this equation strongly emphasises any local variation in the charge density. So if the potentials “look right” then the charge density is also correct. NB. Hex uses a relative permittivity value of 8, instead of the more usual value of around 80. So, in addition to Hex's in vacuo assumption, the numerical values for the potential are likely to differ from other software.
Some molecules may have internal cavities, and these can produce one or more contour surfaces in additional to the primary external surface. Other molecular structures (e.g. structures with many waters, or well-separated domains) may also give multiple surfaces when contoured. As the contouring algorithm implicitly produces positively oriented surfaces (outward normals, positive total volumes), it is convenient to assign each calculated surface to one of two possible classes: Primary (positive volumes, outward normals), or Secondary (negative volumes, inward normals). By default, only Primary surfaces are displayed, but this may be changed using the Draw Surface control.
In addition to the default Gaussian surface, the Surface Type
selector provides the option to draw contoured density functions
of the Sigma and Tau surface functions which are used by
the docking algorithm. These two density functions allow the shape
functions used in docking calculations to be visualised.
Sigma is the external skin density function, typically calculated with
a probe radius of 1.4Å, and Tau is the interior density function
(effectively, the van der Waals volume).
For relatively small molecules, and when using high expansion orders (N),
the Tau density can be seen to give a remarkably good representation
of the initial (atomic) Gaussian density.
The order of the 3D expansion (default N=25) is taken from the
Docking ... Final Search parameter in the Docking Control panel.
Both the Sigma and Tau surfaces are contoured using
a hard-wired density value of 0.25.
However, reconstructing each 3D density from the shape expansion coefficient
vectors is a relatively expensive calculation.
This display mode is mainly intended to illustrate the internal representations
used in docking. Figure 3 shows some example shape density isosurfaces.
In this figure, the highest polynomial power in the SPF expansion, L,
is related to the order N in the Docking Control panel by L=N-1.
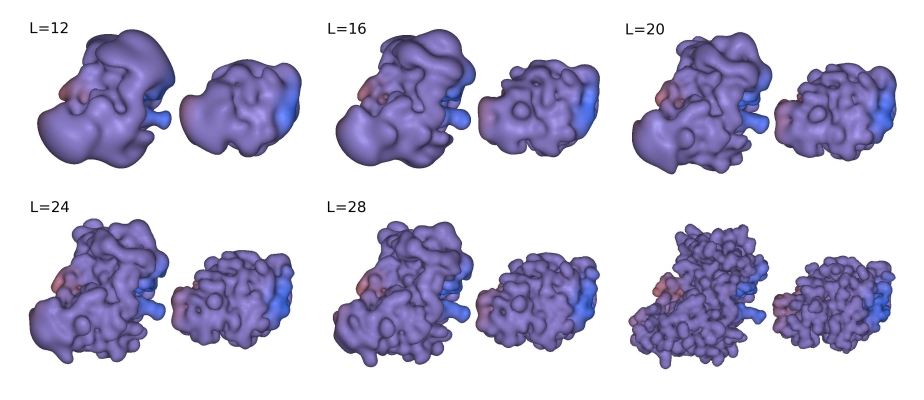
Figure 3. Illustration of the SPF steric density representations at various 3D expansion orders for the complex between the HyHel-5 antibody Fv domain (left) and quail lysozyme (right). From top left to bottom right: steric density isosurfaces shown at expansion orders L=12, 16, 20, 24, and 26, with the subunits separated by 15 Å to give a full view of each domain. The bottom right pair shows the van der Waals surfaces from which the SPF expansions are derived.
The Sigma/Tau Shift slider controls how the shape (and electrostatic) expansion coefficients are computationally translated using the T(R) translation matrices prior to reconstructing the surface from translated expansion coefficients. Thus it is possible to view graphically how the representation used in the docking correlation degrades with increasing distance from the origin. In this display mode, the receptor surface is translated in the negative z direction, and the ligand surface is translated in positive z for ease of viewing. You should see a blurring of those portions of each molecule furthest from the origin, although the shapes of the surface regions near the origin are very well preserved after translation. NB. In Hex's docking correlations, all computational translations are applied entirely to the receptor.
Please be aware that drawing surfaces can use a lot of memory. Drawing the surface of a large molecule using a fine (0.25-0.5 Ångstrom) grid can use hundreds of megabytes of memory. Attempting to draw a complex surface on a machine with insufficient memory could cause the machine to hang.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In Hex version 5.0, spherical harmonic (SH) molecular surfaces are generated from the marching tetragons surface described above. Essentially, all of the vertices from the trangulated surface are first projected onto an icosahedral tesselation of the sphere. If more than one surface point maps to the same tesselation vertex, then the point with the largest radial distance from the origin is selected. Hence, a bounding envelope of sample points is constructed. Any gaps in the tesselation which might remain are “filled in” by interpolating from values of nearby non-null tesselation vertices. Hex doesn't use spherical harmonic surfaces in any of its calculations. It only displays them. However, the commercial ParaFit program (also written by Dave Ritchie) can rapidly superpose and spherical harmonic surfaces and surface properties calculated by the ParaSurf program. Both ParaSurf and ParaFit are available from Cepos Insilico Ltd.. Hex can read ParaSurf SDF files, and it can display the key ParaSurf spherical harmonic surface properties.
The Harmonic Surface control panel controls the way in which spherical harmonic molecular surfaces are calculated and displayed. You can view the molecular envelope by selecting:
Graphics ... Harmonic Surface ... Enable Surface (toggle on) |
The icosahedral sampling resolution is controlled by the Mesh Order slider. The order of the spherical harmonic expansion can be selected using the Order (L) slider in the Harmonics Control panel. The SH envelope may be displayed in different styles and colours using the Display Mode and Line Colours selectors. Figure 4 shows the SH surfaces for the antibody/lysozyme example, obtained by pressing the Harmonics short-cut button on the right hand border.
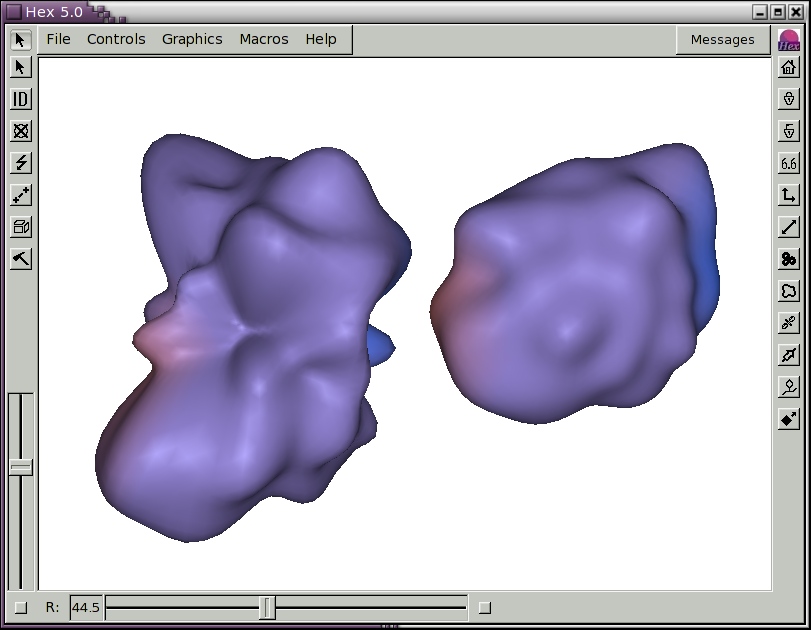
Figure 4. Illustration of spherical harmonic surfaces to order L=12 for the HyHel-5 antibody Fv domain (left) and lysozyme (right).
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Dot surfaces are somewhat of a historical remnant in Hex. They used to be used as an approximate but fast way to calculate the molecular surface, the solvent-accessible surface and the van der Waals surface for docking. However, docking and superposition calculations now use a much more accurate method based on contouring Gaussian density functions to calculate these two surfaces. Nonetheless, it is still sometimes useful to be able to draw a dot surface because Hex uses all the atoms from an input PDB or SDF file to draw the surfaces. Hence dot surfaces may be used to verify the presence of atoms that might not be recognised or drawn properly in other drawing modes. Like the SH surfaces, dot surfaces are now calculated directly from the marching tetragons surface, as described above.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Normally, rotation and scale (zoom) operations are applied relative to the scene origin. When just one molecule is loaded, the scene origin coincides with the centroid of the molecule. However, when two molecules are loaded, the scene origin is taken as the midpoint between the two molecular centroids. This behaviour can be changed by using the first selector in the Orientation Control panel to select a given molecular centroid or a specific atom to act as the origin. The Select Origin button (a circle with a diagonal cross through it) on the left hand border may also be used to select the zoom/rotation centre.
Although docking calculations normally move both the ligand and the receptor, the default display behaviour is to keep the receptor fixed in space and to assign all the motion to the ligand as successive docking solutions are viewed. This behaviour can be changed using the Docking Motion selector in the Orientation Control panel.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The Clipping button (three faces of a cube) on the left hand border selects scene clipping mode. You can define up to six clipping planes in the scene, several of which may be moved together using a single slider. However, controlling clipping is probably the most difficult part of the program to master, since this involves the use of all three mouse buttons and several keyboard keys, in addition to the basic graphical widgets.
When scene clipping mode is selected, you should see a wire frame drawing of six skeletal clipping planes centred in the scene. A new slider control is also drawn in the bottom right border. This new slider may be used to move selected clipping planes through the scene, in plane-perpendicular directions.
In clipping mode,
the mouse buttons may be used to translate and rotate the clipping
skeleton in the usual way. Note however, that the rest of the scene remains
fixed. You can use the keyboard Space Bar to toggle between moving the
skeleton about the scene and moving the scene about the skeleton.
Picking one of the skeletal planes
(using Button 1 for picking) selects that plane and
attaches it to the scene.
The plane is now drawn in white,
either as a filled shape or as an outline,
depending on which face you happen to be viewing.
The mouse buttons now move only this plane relative to the scene (or, if
you have hit the Space Bar,
the mouse moves the plane and the scene together).
The filled face will become the clipping face of that plane.
Button 2 may be used to activate
the selected plane (just press Button 2 anywhere in the scene). That is,
every part of the scene above the filled face should become clipped. Pressing
Button 2 again toggles clipping. Try moving the clipping plane though
the scene using the right slider,
or by using the mouse and the Space Bar to control the movement.
Try using the Delete Key (keyboard backspace)
to toggle the sense of the clipping plane. Once you are happy with the
position of the clipping plane, you can revert to Pointer Mode
(select the Left Arrow button) or you can proceed to pick and
activate further clipping planes.
In pointer mode, the right hand slider continues to operate the
most recently activated clipping plane.
It is also possible to make the slider move two planes simultaneously.
Having activated a clipping
plane, now try picking it with Button 3. This draws the plane in yellow
and ties its motion to the slider. Now pick and activate the clipping plane
(using Buttons 1 and 2) on the opposite face of the clipping skeleton.
The right
slider should now move both planes together through the scene. You can tie
multiple planes to the slider, although selecting two perpendicular planes
is probably the most sensible option. As you might expect, picking
a plane with Button 3 for a second time unties it from the slider.
At any time while in clipping mode, you can undo the most recent operation using the Hammer button. Three successive clicks on the Hammer will clear all clipping settings and will return the display to an unclipped scene.
If necessary, you can use the keyboard Plus and Minus keys to
zoom the clipping skeleton independently of the scene.
If the scene contains molecular surfaces (dots, lines or polygons),
you can use the keyboard Equals key to toggle between clipping both
the molecule and its surface (the default) and clipping just the surface,
leaving the molecular skeleton fully visible.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The Animation Control panel allows you to run a “movie” of either the results of a docking calculation, or a sequence of models from an NMR structure, for example, and it controls how the scene “spins” if you perform a mouse drag-release action.
After a
docking run, pressing Start in the Graphics ... Animation
control panel will cause Hex to draw each docking solution in turn.
The rate at which orientations are drawn is controlled by the
Frame Rate slider. Similarly, if you have loaded a PDB file
that contains several NMR model structures, setting the
Movie Type to Receptor Models or
Ligand Models, as appropriate,
and pressing Start will show a movie of the sequence of models.
The orientations, or frames, of both types of movie may be shown just
once, or cycled forever. The movie can be stopped at the current
frame by pressing the Stop button. The speed of the movie is
controlled by the Frame Rate slider.
The maximum frame rate achievable will depend on the speed of the CPU and on whether your machine has hardware-accelerated graphics. The Frame Rate thus defines a requested rate. Actual performance will vary. But please note, Hex never hogs the CPU in a tight loop on input events, even during an animation, as do many other programs (which shall remain nameless). Thus it should still be possible to perform other activities while a movie is running without things becoming sluggish.
You can spin the scene by
dragging with Button 1 or Button 2
to start a rotation and by releasing
the button while still moving the mouse to initiate a continuous rotation
(spinning).
When spinning, the rotatation angle is incremented by the
current value in the Spin Angle slider, and new rotational increments
are drawn at the current Frame Rate. If desired, the mouse button
action that initiates spinning can be enabled/disabled with the
Enable Spinning toggle. Spinning is enabled by default, and it is
possible to spin a movie (if thats what you really want to see!).
Any of the usual user interface controls may be used while an animation is running. If using these controls changes the graphical complexity of the scene, Hex will take a second or two to adjust the drawing speed back to the requested frame rate.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Personally, I think full-screen mode is the best way to view molecular
graphics scenes. You can press F to switch back and forth
between full-screen and window-manager modes. By default, Hex starts
in window-manager mode.
You can also press G
at any time to toggle the display of the GUI controls when in full-screen mode.
G only toggles the setting, not the display. So you won't see the
effect until you enter full-screen mode.
In both the Linux and Windows versions, you should always use F to
get full-screen mode because this forces the window border off.
Conversely, don't use the window manager's “maximise” control because
this forces the window to keep a border, and F won't then work.
In Windows XP, you will probably still see the Windows Taskbar in
full-screen mode. To get rid of this, open the
Taskbar and Start Menu in the Windows XP Control Panel and
uncheck the Keep the taskbar on top of other windows item. I don't
know whether this feature is available on earlier versions of Windows.
In Mac OS X, full-screen mode does not override the Mac Taskbar and Dock panels. Hence it is best not to use Hex's full screen mode, but instead use the window manager's “maximise” button to ensure the graphics window is well-behaved.
In Linux, full-screen GUI mode with the Gnome window manager
is “quirky”. Everything works as it should, except that Hex
control panels can't be re-raised over the full-screen window after the
control panel loses mouse focus (i.e. when the cursor leaves the panel).
You will need to revert to window-manager mode (F) to see everything
again! These problems seem to be due to a strange interaction between
the Gnome window manager and the FLTK interface toolkit.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Stereo is known to work on Windows XP and RedHat Linux 8.0 (kernel version 2.4.18-14, XFree86 version 4.0.0) or later with an nVidia Quadro4 XGL700 card, and using a recent nVidia driver (1.0.3123 or later). For best results with Hex and nVidia cards, please use the latest versions of both driver software, service packs, and operating systems. The recent nVidia drivers give Hex both stereo-in-a-window and full-screen stereo on both Windows XP and Linux. nVidia's 4351 driver (May 2003) is excellent, and is very easy to install. See section nVidia Stereo Configuration for more details. However, if you are upgrading an nVidia driver on Windows, ensure you de-install any earlier drivers first.
The type of graphics visual that Hex uses is determined
when the program first starts up. By default, Hex tries to use
a stereographics window. However, this nearly halves the drawing
rate compared to non-stereo drawing. So if you don't like/want stereo,
you can force stereo off by using hex -nostereo.
Alternatively, set the HEX_STEREO environment variable to a
false value (i.e. any one of: no, n, false, f, or 0) in your login script.
If a stereo visual has been selected,
the display may be toggled between stereo and mono using the
Stereo Parallax option in the
Projection Control panel
(or simply by pressing the keyboard S key).
The Parallax
slider may be used to adjust the stereographic effect.
Stereo parallax is the perceived separation
between the left and right images in the viewing plane (monitor screen).
The numerical values in the parallax slider
correspond approximately to screen millimetres on an un-zoomed display.
Positive parallax
values make the scene appear “inside” the display, whereas negative values
cause the scene to appear through, or in front of, the plane of the display.
A better stereographic effect is achieved when the
Perspective Projection toggle is enabled (by default, an orthographic
projection is used).
The keyboard P key may also be used to toggle
between perspective and orthographic projection modes.
When perspective is enabled, a checker-board background may also be enabled
to enhance the perspective effect (keyboard B).
Figure 5 shows the antibody/lysozyme scene with perspective and background
modes enabled.
The perspective effect can be
increased by reducing the Far Plane and/or increasing the Near
Plane slider values.
For most people, a small positive parallax and a moderate perspective
gives a pleasing effect without straining the eyes.
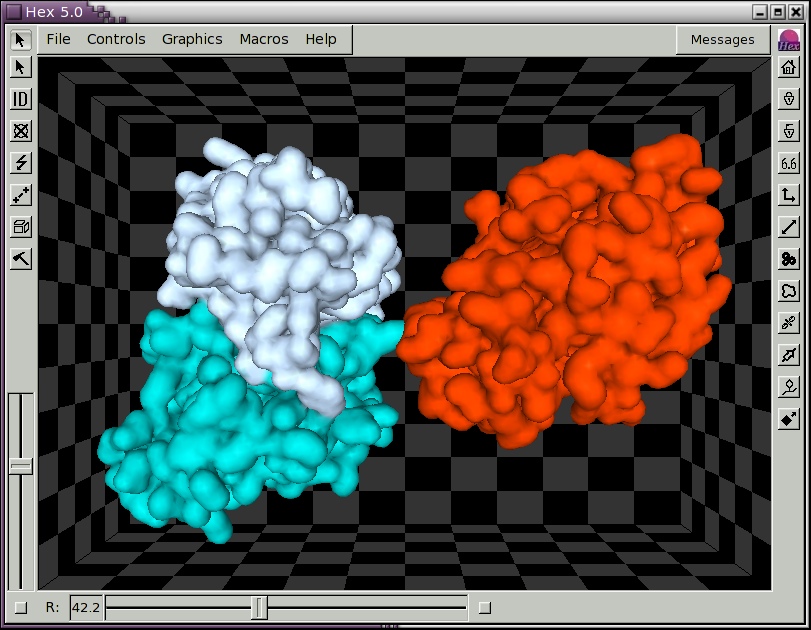
Figure 5. Illustration of the HyHel-5/Lysozyme complex shown as contoured Gaussian density surfaces and coloured by chain colour, drawn using perspective (keyboard P) and background (keyboard B) modes enabled.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
When Hex reads in a molecule, it uses its all-atom centre of mass as its centroid. This centroid is used as the local coordinate origin for docking, and as the point about which any molecular rotations are applied. However, if one or both molecules are quite large (i.e. too big to fit within about a 30 Ångstrom radius ball), you will probably want to change the centroids used for docking. This is because Hex's radial functions fall off rapidly beyond about 30Å from the origin. Hence, unless you move the origin(s) to be closer to the expected binding site(s), you are likely to get very poor docking results. If you don't know or suspect where the binding site is on a very large molecule, you should use Hex's macro docking mode for such cases. See section Docking Very Large Molecules (Macro Docking).
In addition to rotating, translating, and scaling a molecular graphics scene, Hex, also allows you to change the relative orientations of a pair of molecules. Hex calls this editing, because saving “manoeuvred” structures out to a file causes them to be written using their transformed coordinates. Note: Edit Mode is only enabled when you have two molecules loaded. Editing is often a necessary preparatory step before running a docking calculation. For example, when you load a pair of molecules, Hex uses some heuristics to place the ligand near the receptor. However, you may have knowledge about one or both of the binding sites, and hence you may want to manoeuvre the molecules into something that resembles the expected binding orientation. If you do this, you can then limit the search range of the docking correlation instead of performing a global search. This should give fewer “false-positives”, and it will certainly make the docking calculation go faster.
It should be noted that although you can edit the scene by picking and moving both the receptor and the ligand orientations, the coordinates of the receptor “take priority” when structures are written to an output file. In other words, any relative motion is always transferred entirely onto the ligand on output. The receptor coordinates are always restored to their original values.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Molecular centroids may be edited using the Edit Origins button
(two crosses and a line) on the left-hand border.
When editing orientations,
it may help to activate the Intermolecular Axis,
in order to see the new and old positions of the centroid(s).
Try changing to Edit Origins mode, and pick a molecule to edit.
Once the picked molecule is highlighted, dragging Button 1 will
move the origin of the selected molecule.
In Edit Origins mode, buttons 2 and 3 always rotate the entire scene.
When you revert to the default
Pointer Mode, the new origin will be activated.
Note that the new origin only applies
to the current session. It is not saved when you write an edited molecule
to disc.
You can use virtually all of the other GUI functions while in edit mode,
although certain operations
(e.g. opening/closing molecules) cause Hex to revert to pointer mode.
You can also explicitly specify the location of the molecular origins using
the Receptor Origin and Ligand Origin text entry boxes
in the Orientation Control panel.
For example, following the HyHel-5/lysozyme example,
you could enter Y-38 into the Ligand Origin text box
(and then press the return key) in order to set the ligand origin to the
the CA atom of PHE:38 in the lysozyme Y chain. Similarly, as of
Hex version 5.0,
the Receptor Interface and Ligand Interface
text entry boxes may be used to specify the residues which should be located
on the positive or negative z-axis, respectively.
Orienting each protein with respect to the z-axis in this way
is equivalent to specifying initial values for five of the six rigid body
degrees of freedom (i.e. all but the twist angle).
Another way to control the molecular orientations is to use the Apply to selector in the Orientation Control panel to select either the receptor or the ligand, and then to rotate the selected molecule using the Euler angle Alpha, Beta and Gamma sliders. The intermolecular separation can be modified with the R slider at the bottom of the main window. Select Commit when you're done editing, in order to commit the new transformations into the molecules.
Orientation editing can also be performed manually clicking on the
Edit Mode left border button (which looks like a lightning bolt)
and then by picking and dragging molecules using the mouse.
In both editing modes, picking a molecule
ties its motion to the mouse and to the rotation sliders in the
Orientation Control panel.
The covalent skeleton of the picked molecule is displayed as
dashed lines as a
visual reminder that this molecule has been activated for editing.
Picking the background (i.e. a Button 1
pick that misses all atoms) de-selects the active molecule
and makes the mouse move the whole scene.
The Hammer button can be used to undo the most recent sequence
of editing movements.
Once you're happy with
your edits, revert to Pointer Mode to commit the new orientations.
You can now save the
scene by going to the File menu and selecting:
File ... Save ... Both |
This causes a file selection widget to appear, prompting you for the name of a file in which to save both molecules (in the newly edited orientation). You can confirm the new orientation has been created by loading the new file into another Hex session (you may have to rotate the scene to see exactly the same view). Alternatively, you could write each molecule to a separate file using:
File ... Save ... Receptor |
and
File ... Save ... Ligand |
You could then open these new files to verify the new orientation.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In order to run a docking calculation in Hex, you will need to load a receptor and a ligand PDB structure using the File pull-down menu. If you want to test the docking algorithm by docking two separately determined sub-units of a complex for which the crystal structure is also available, you can also load the complex structure which will be used as a reference orientation to evaluate the accuracy of the docking prediction.
Generally, you will have to remove water molecules and any other hetero molecules prior to docking. You can do this globally using the Hetero Control menu panel. If more detailed control is required, you will probably have to edit each PDB file manually using a text editor. It may also be necessary to remove other chains in the PDB file or to shorten a chain to the domain of interest in docking. For example, when docking an antigen to an antibody it is usually advisable to delete all but the Fv fragment of the antibody structure (although the program has been used to dock a protein G molecule to a complete Fab fragment). Having edited your PDB files, you should have a receptor and a ligand file which contain only the receptor and ligand molecules, respectively, and (optionally) a complex file, which contains both molecules in the docked orientation. When using a complex structure, you should ensure that the chain names and residue numbers are consistent with those of the receptor and ligand because Hex uses this information to identify and hence superpose corresponding pairs of alpha-carbon atoms from each chain in order to calculate RMS deviations between the docked position of the ligand and its position in the known complex. If necessary, you can use the Linux utility hex_chain in Hex's bin directory to rename a chain in a PDB file. Similarly, the hex_delta utility may be used to renumber the residues of a given chain.
Up to three protein structures can be loaded into Hex directly from the command line. For example, to load three of the PDB files from the examples directory for the HyHel-5/lysozyme complex, you could type at the Linux command prompt:
hex 3hfl_fv.pdb 3hfl_ly.pdb 3hfl.pdb |
If you are studying a particular system and you find you are continually repeating the same sequence of actions to get things set up, you may find it to be more convenient to load all three molecules together using a short macro file, similar to those in the examples directory. For example, the macro file, 3hfl.mac, can be executed by selecting:
Macros ... Run ... 3hfl.mac |
This macro file contains the following:
# 3hfl.mac # ======== # close_all set_colour_file 3hfl.col open_receptor 3hfl_fv.pdb open_ligand 3hfl_ly.pdb open_complex 3hfl.pdb fit_ligand |
When using a complex reference structure, you should find that on loading the molecules, the complex is drawn in grey and the complex molecule is superposed onto the receptor (it is the complex that physically moves here). If the ligand molecule originated from an edited complex file, then it will also be superposed over the complex because its coordinates are relative to the same coordinate frame as the receptor molecule. Otherwise, the ligand can be transformed into the receptor/complex frame using the Fit Ligand button in the Orientation Control panel.
Note. If you wish to superpose one or more proteins onto another, the above behaviour may be exploited by treating the stationary molecule as the “receptor,” and by treating each moving molecule as the “complex.” Saving the fitted “complex” to a new PDB file will effectively write it out its fitted orientation in the coordinate frame of the receptor.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Docking calculations are controlled by the options in the Docking Control panel. Generally, the docking search proceeds by rotating the receptor and ligand about their centroids at each of a range of intermolecular distances. The receptor and ligand are each assigned two Euler rotation angles, and the final rotation is defined as a twist of the ligand about the intermolecular axis. The default behaviour is then to perform a full six-dimensional search over the full rotational ranges. However, more limited docking searches can be performed in which the allowed angular and distance ranges may be constrained by the user. The docking coordinate system is illustrated in Figure 6 below. See section Manoeuvring Molecules for details on setting up the initial docking orientation.
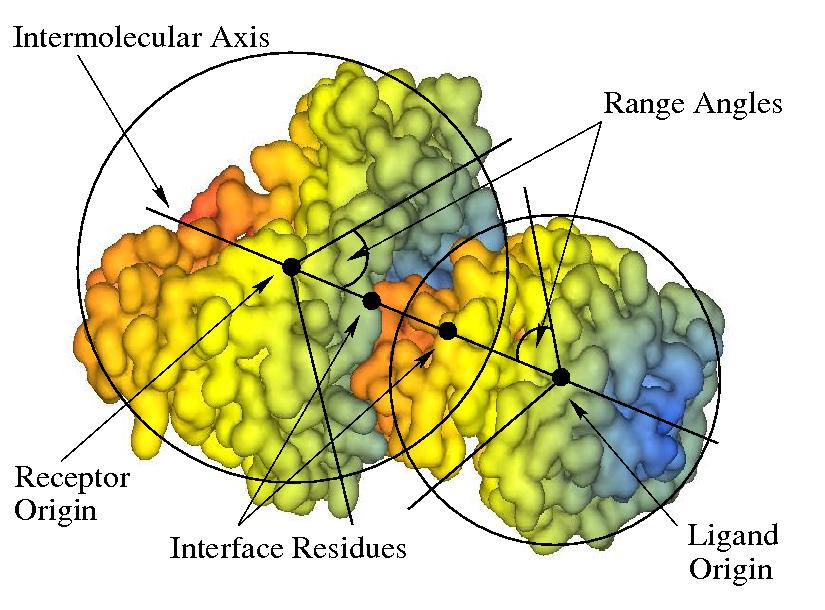
Figure 6. Illustration of spherical polar docking with respect to the intermolecular axis. An initial docking orientation may be defined by specifying which residues should be located at the local coordinate origin for each molecule, and by defining "interface residues" which will be located on the z-axis. The docking search may be restricted by defining a “range angle” for the receptor and/or ligand orientations. If range angles are defined, then the interface residues will always be constrained to appear within a spherical cone defined by the corresponding range angle. This illustration shows two range angles, each of 45 degrees.
The calculation is arranged so that the intermolecular twist angle search is in the innermost loop of the search. The search around the twist angle may be accelerated using a 1D FFT. Alternatively, all three Euler angles assigned to the ligand can be searched together using a 3D FFT. In the Linux version, all five rotation angles may be searched together using a 5D FFT. However, this requires at least 1 gigabyte of memory to hold the very large 5D rotational grid.
Note. The speed of the 3D and 5D FFT calculations depends very much on the quality of FFT code, and whether it exploits any hardware acceleration. For CPU-based docking, the Intel MKL code is about twice as fast as FFTW, which is about one and a half times as fast my own multi-dimensional FFT code which is partly based on Kiss FFT, and which doesn't use any assembly or SSE instructions. However, only the Linux version of the MKL library is available for free to non-profit organisations. So docking is fastest with the Linux version of Hex. I do not use the FFTW code because I do not agree with its GPL license conditions. Hence, 3D and 5D docking correlations on the Windows and Mac versions of Hex is relatively slow.
For GPU-based docking, 1D correlations using cuFFT, on a high-end GeForce or Quadro card are much faster than on a high-end CPU. Compared to using a single high-end CPU (3.2GHz i7-965), the same calculation on a high-end GPU (FX-5800) is about 45x faster (Ritchie and Venkatraman, manuscript in preparation).
There are several controls which specify the resolution, and in particular the order, N, of the docking correlation. Figure 7, below, shows a screen shot of the Docking Control panel. The default settings are for the program to perform an initial Steric Scan at N=16, followed by a Final Search at N=25, using just the steric contribution to the docking energy. In this mode, about all but the top 30,000 orientations are discarded after the Steric Scan. The Steric Scan may be toggled off, in which case every orientation is evaluated using a steric correlation (and optionally an electrostatic correlation) to order N, as given the Final Search slider. However, this can significantly increase total docking times. Using the two-step search with N=16 and N=25 is found to work well in practically all cases. The electrostatic contribution to the docking correlation may be enabled using the Electrostatics toggle. Electrostatics are only ever calculated in the Final Search phase. You can get a feeling for how well each correlation order, N, recognises a given complex by running the program on known complexes and by observing how highly the correct solution is ranked in each case.
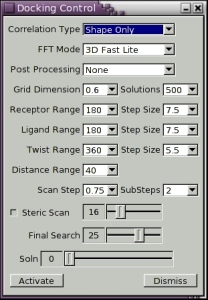
Figure 7. Screen shot of the Docking Control panel.
Although the default is to use correlations to N=25, in Rounds 3–5 of the CAPRI blind trial, I found that better results are obtained using N=30 correlations (see section References). Generally, N=30 is recommended when docking high resolution crystal structures for which the conformational change on binding is expected to be small. N=25 should be used when docking model-built structures or structures which are expected to be more flexible. As rule of thumb, I would use N=16 for the scan stage with final scoring at N=30, and I would use scans using N=20 when scoring with N=30. In some cases, a good solution can be missed with the N=16 scan. It is safer, but obviously slower always to use N=20 for the initial scan.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
As of version 4.5, Hex performs the high resolution Final Search correlation using smaller distance incerements than are used for the fast low resolution Steric Scan phase. This allows the search space to be covered more rapidly (coarsely) in the first phase, but more finely in the final phase. This behaviour is controlled by the Distance Range, Scan Step and SubSteps parameters in the Docking Control panel. The default values are Distance Range=40, Scan Step=0.75, SubSteps=2. This means that the Steric Scan phase will search over 55 distance increments of +/- 0.75 Å from the starting separation, plus the starting separation itself). These orientations are sorted by calculated energy, and a new set of trial orientations are generated for the top-scoring 10,000–20,000 orientations using the Scan Step and SubSteps parameters to construct new distance samples in steps of +/-(Scan Step)/(Substeps) from the initial orientations. In other words the default behaviour is essentially to scan the search space at 1Å resolution, but to perform the high resolution scoring at 0.5Å resolution. Setting SubSteps=0 gives the old behaviour of earlier versions in which a constant distance step is used for both resolution levels.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
OK, lets start docking. Open the Docking Control panel and select the following (mostly default) values from the selection boxes:
Controls ... Docking ... Correlation Type ... Shape Only Controls ... Docking ... FFT Mode ... 3D Fast Lite Controls ... Docking ... Post Processing ... None Controls ... Docking ... Grid Dimension ... 0.6 Controls ... Docking ... Solutions ... 500 Controls ... Docking ... Receptor Range ... 45 Controls ... Docking ... Receptor Step Size ... 7.5 Controls ... Docking ... Ligand Range ... 45 Controls ... Docking ... Ligand Step Size ... 7.5 Controls ... Docking ... Twist Range ... 360 Controls ... Docking ... Twist Step Size ... 5.5 Controls ... Docking ... Distance Range ... 40 Controls ... Docking ... Scan Step ... 0.75 Controls ... Docking ... Substeps ... 2 Controls ... Docking ... Steric Scan ... 16 Controls ... Docking ... Final Search ... 25 |
This says that the Steric Scan (N=16) phase of the docking calculation will be performed at (1+40)/0.75=53 intermolecular separations, in +/- steps of 0.75 Å starting from the current distance posted in the R slider in the bottom border of the main window. The Final Search (N=25) phase will be applied to the the highest scoring scan orientations in steps of 0.75/2 Å, as described above. The rotational search will use angular increments of about 7.5 degrees in each of the two ligand and receptor rotational angles, and in steps of 5.5 degrees about the twist angle. Internally, the given step sizes will be adjusted to give computationally convenient numbers of samples. For example, a step size of 5.5 degrees will actually give 64 samples (360/64=5.625). Specifying a step size of 7.5 degrees will give 48 or 24 angular samples in the corresponding FFT grid, or it will select an icosahedral tesselation with 812 vertices because that is the tesselation order which will give an average angular distance between neighbouring pairs of vertices of about 7.5 degrees.
Note. This method specifying the angular resolution is new in version 5.0. Earlier versions required the user to specify the sampling density directly in terms of tesselation orders, which was not very easy to understand! Please also note that the default search step sizes are often somewhat finer than the search increments used in previous versions.
Now start the calculation:
Controls ... Docking ... Activate |
You have just started an example docking search restricted to the known binding sites, so the calculation should only take a few minutes. The program will take a few seconds to calculate the surface skin coefficients and then proceed to calculate docking correlation scores at each of the specified angular and intermolecular increments. A Cartesian grid is used to sample the molecular skins numerically but this grid plays no further role in the calculation once the surface skin expansion coefficients have been determined. Most conventional FFT docking algorithms have to use rather large grids (e.g. 1 Ångstrom cubes) because the grid must accommodate all possible translations of the ligand about a stationary receptor. Here, the grid only needs to contain the larger of the two molecules so that much finer sampling grids are feasible. In Hex, a 0.6 Ångstrom grid seems to work well and is still reasonably fast to calculate. The sampling grid size may be varied using the Grid Dimension selection box. The calculation of the surface skins used in the docking correlation is controlled by the parameters in the Surface Control panel. The default values do not normally need to be changed.
The search in the above example is “restricted” because setting the ligand and receptor range angles to 45 degrees means that only a small fraction of the total possible rotational increments (the Receptor Samples and Ligand Samples values) will actually be used for the molecular rotations of each molecule. Essentially, each molecule is rotated incrementally so that successive tessellation points are rotated onto the intermolecular axis (the z-axis). Thus, in the low resolution Steric Scan phase, 812 x 812 distinct rotational orientations are produced, although any orientations that fall outside the angular range cones are discarded. Angular search ranges are illustrated in Figure 6. At each of the surviving orientations, the ligand is then rotated (or twisted) through 360 degrees about the z-axis in 64 steps of about 5.5 degree increments specified by the Twist Samples value. In the high resolution Final Search phase, the best 10,000 of the above orientations will be used to generate up to 25,000 new distinct trial orientations after distance sub-sampling (see above) and re-evaluated at N=25 using 128 samples for each twist angle search. Note, Hex is hard-wired to use 64 twist steps in the initial steric scan phase, because there is no benefit in using finer steps in this phase.
It should be noted that such docking calculations never give a unique solution. Rather, Hex sorts the generated orientations by docking energy and prints a summary of the 10,000 highest scoring (lowest energy) orientations. The best 500 orientations are retained for viewing. You can view the solutions using the Soln: slider, or you can step through the solutions using the keypad or arrow keys:
Home: Starting orientation Page/Down: Next solution Page/Up: Previous solution End: 500'th (or last) solution |
You should find in the above example that Hex generates several orientations with low RMS deviations from the correct (starting) orientation. However, you might object that the calculation was biased to find the right answer by restricting the search to the known binding sites. If so, you could try a global docking search (about 10^9 trial orientations), using:
Controls ... Docking ... Receptor Range ... 180 Controls ... Docking ... Ligand Range ... 180 |
This calculation will take about 30 minutes on a single 1GHz Pentium III processor. This is substantially faster than an equivalent calculation using a conventional FFT approach. In this particular case, Hex should still identify the correct docking orientation (to within about 1.5 Ångstrom RMS), but its often useful to use restricted searches when starting with unbound subunits.
When viewing docking solutions, it is helpful to keep either the receptor or the ligand in a fixed orientation in the display, so that only one molecule moves when stepping through the docking solutions. The default is to transfer any receptor motion onto the ligand, so that the ligand appears to translate and rotate about a fixed receptor. This behaviour can be changed using the Orientation Control panel:
Controls ... Orientation ... Centre ... Ligand |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
If you plan to use Hex more than once, enabling a disc cache will speed up the translational part of each calculation. When disc caching is enabled the (large) translation matrices are calculated only when necessary and are saved in the cache directory for re-use. Disc caching controls can be found under the File menu. The default cache directory is $HEX_ROOT/cache, although it is recommended to set a cache directory explicitly using the HEX_CACHE environment variable. The cache directory should be specially created for this purpose (i.e. it contains nothing else) and a distinct directory should be used by each user (because there's no synchronisation between multiple invocations of Hex). With typical usage, the disc cache will occupy up to about 200 Mb of disc space.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Following the basic docking correlation algorithm, candidate docking orientations may be filtered and refined using one of the Post Processing options. Post processing is applied to the top-scoring 1,000 solutions from the correlation search (or the user-selected number of orientations to retain, if larger than 1,000). The simplest option is to enable a bumps counter, in which the number of steric clashes between non-bonded pairs of heavy atoms in each solution is calculated. An option in the Clustering Control panel may then be used to filter out solutions with a specified number of steric clashes.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In addition to the bumps counter, a single (rigid body) molecular mechanics energy may be calculated for each docking solution (MM Energies), or a Newton-like energy miminisation (MM Minimisation) can be applied to each docking solution. These energies are calculated using “soft” Lennard-Jones and hydrogen bond potentials, adapted from the OPLS force-field parameters, along with an explicit charge-charge electrostatic contribution. When docking complexes where conformational changes are known to be small, this gives an effective way to prune many “false-positive” orientations and to enhance the energy of the “right answer”. However, this rigid-body refinement procedure should not be used if conformational changes are expected to be large because (despite using soft potentials) it tends to eject ligands with incorrect conformations from the binding site. This part of the program is still “under development”.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Because Hex uses essentially a brute-force search approach to the docking problem, it is advisable to over-sample the search space rather than to risk missing a good solution by under-sampling the space. However, this can cause multiple similar but incorrect orientations (false-positives) to push good solutions down the list. By default, Hex uses a simple clustering algorithm to group spatially similar docking orientations. Each docking solution is first ordered by energy, and the lowest energy solution is made the seed orientation for the first cluster. The list is then searched down to a given depth for other similar orientations whose main-chain alpha-Carbon RMS deviation is within a given threshold (default 3Å RMS) of the seed orientation, and these orientations are then assigned to the first cluster. The process is then repeated starting from the next lowest unassigned orientation, until all solutions have been assigned to a cluster. The Cluster Window parameter may be used to control the search depth when looking for cluster members. Because clustering uses a simple but inefficient algorithm (rather like a “bubble-sort”), it is advisable use this parameter to limit the search depth if the number of saved solutions is large.
The Controls ... Clustering control panel may be used to control the
type of clustering and clustering parameters used, and how
cluster membership affects the displayed solutions. By default, only the
lowest energy member of each cluster is displayed or written to an output
PDB file (Display Clusters = Best).
Alternatively, you can choose to
view (or output) all solutions (Display Clusters = All).
Similarly, solutions
may be viewed in cluster order (best to worst) or in order of calculated
energy using the Sort Solutions selection option.
The RMS threshold for cluster membership may be changed using the
RMS Threshold slider.
Displayed solutions may also be selected by
the number of steric clashes, or bumps, detected. Any solution containing
a number of bumps which is less than or equal to the current
Bumps Threshold (default 0) is a candidate for cluster membership.
Solutions with a greater number of bumps may still be viewed or output,
but these always appear after any non-bumping solutions in the sorted
list of solutions (i.e. have a larger cluster rank).
The default docking search range and clustering parameters are normally sufficient to generate good coverage of the search space, and to distinguish different but similar solutions. But you may wish to experiment. Clustering is re-calculated whenever a clustering parameter is changed, without having to re-run the docking search. Here, “good coverage” means that for each docking orientation, there is about a 99% chance of generating at least one other solution within 3Å RMS of the first one (unpublished experiments). Deciding whether or not two similar but different orientations ought to fall within the same cluster is somewhat subjective. The 3Å threshold seems to work well for large-ish ligands (roughly 100 residues), but should probably be reduced for smaller ligands. In the CAPRI blind docking experiment, I tend to use a large threshold of, say, 5Å to try to improve the chances of finding at least one “medium accuracy” prediction within the first 10 predictions submitted). But this doesn't always help!
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
There are two main ways in which docking results may be saved to disc.
The most compact method is to write
a docking summary file using the File ... Save ... Transform
menu item. This writes a file that lists the docking energies for all
predicted orientations, along with the rotation and translation parameters
which should be applied to the ligand coordinates in the original ligand
PDB file in order to produce the corresponding docking orientation in the
coordinate frame of the recptor PDB file. The actual file format is descibed
in the first few “comment” lines of the output file.
A similar but more detailed
output file format is given by File ... Save ... Matrix. This
lists the transformations in a more verbose format using keywords to
label each piece of data.
For rigid body docking, both formats are quite compact,
but you will need to write your own code to
process the docking summary file.
Its not possible to read these files
back into Hex; they are purely for output to external software.
The second, less compact output method is to write a separate PDB file for each docking orientation. The current docking orientation can be written to a single PDB file by selecting:
File ... Save ... Both |
Alternatively, a range of docking solutions may be saved using:
File ... Save ... Range |
This opens a new Save Docking Range panel in which a range
of docking solutions may be specified, along with a
a file name pattern to be used for the output files.
The default is to save the first 100 solutions to
separate PDB files of the form dock001.pdb, etc., in the current
directory.
In both the PDB range and the summary output modes, either a
list of raw solutions (sorted by energy) are written, or a range of
clustered orientations are written, depending on the current clustering
option. In other words, the sequence of solutions written out is the
same sequence that you would see when stepping through the orientations
using the Page/Down and Page/Up or arrow keys.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Docking multiple structures (e.g. NMR structures) with Hex is easy.
Just put all your ligand structures (assumed to be in a
common coordinate frame) into a single PDB file, and use
the PDB convention of MODEL/ENDMDL to distinguish each structure.
Hex will run its docking search for each ligand in turn. Similarly,
multiple receptor model structures may be loaded and docked, giving an
M by N docking (may take a while though!). The actual structures
to be docked may be selected using the Receptor Docking
and Ligand Docking selectors in the Controls ... Solid Models
control panel (default all v's all). Individual structures may be
selected for viewing using the Receptor Display and
Ligand Display selectors in the same control panel.
This panel also contains some
experimental controls for cloning and editing different models
(where each clone is treated as a new NMR-type model structure
to be docked, with each docking run each starting from the edited pose).
Hex can also read a list of ligands from a multi-molecule SDF file. However, docking results are always written out in PDB format.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The radial shape functions used in Hex decay exponentially beyond
about 35 Ångstroms from the chosen origin. This means that the shapes
of proteins much larger than this are not represented well.
One example of such a large protein is the viral-surface
hemagglutinin molecule presented in the CASP II and CAPRI
protein docking challenges. In order to dock such large molecules,
Hex assigns
multiple local coordinate systems to the larger molecule (assumed to
be the receptor) and docks the ligand around each local coordinate
frame on the receptor. Setting up
the receptor coordinate systems is controlled by the
Controls ... Macro-Sampling control panel.
The general approach is to construct a very low resolution spherical harmonic surface representation of the receptor (say, setting L=5 in the Harmonics Control panel), and then to cover this surface with a large number of spheres. These spheres are placed using the surface normals of each triangular patch used to draw the harmonic surface. The spheres are then iteratively culled, where the sphere with the greatest overlap with its neighbours is culled at each iteration. This is repeated until some pre-set number of spheres (default 25) remain - these should still cover the surface, but be reasonably well-dispersed. The positions of these spheres are then used to generate initial docking orientations for the ligand over the receptor, and an appropriate local coordinate frame is derived from the geometry. For multi-domain receptors, each sphere can be assigned to a source chain, and spheres that do not belong to a given chain may optionally be excluded. The gives a simple (but imperfect) way to limit the docking search to one of several symmetry-related domains (as in the hemagglutinin case, for example).
In order to set up such a calculation for an antibody/antigen system (where the antibody's binding site is known), it is a good idea to orient the ligand (antibody) such that the binding site faces the receptor - i.e. the principal intermolecular axis passes near the antibody hypervariable loops. This can be achieved by manually editing the antibody into position. Then, the macro-sampling algorithm will place the ligand (antibody) at each of the generated starting orientations and in each orientation the antibody will be transformed to face the receptor. Using the main Docking Control panel, a search range of 45 degrees should be set for both the ligand and receptor so that the docking search will gyrate each molecule about the local intermolecular coordinate system yet still keep each molecule roughly facing the other as defined by the starting pose.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
There exist a few further parameters which control the docking calculation but which are not accessible from the GUI. These parameters must be set using keywords in a macro file. For example, the steric overlap penalty factor has a default value of 11. This can be adjusted by running the example macro factor_q.mac:
# # steric_q.mac - change steric penalty factor Q (default=11). # # (this parameter can't be changed from the GUI yet). # factor_q 11 |
Similarly, another example macro file is factor_g.mac:
# # factor_g.mac - change electrostatic scale factor (def=700 J/mol). # # (this parameter can't be changed from the GUI yet). factor_g 700 |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex superposes or matches pairs of molecular structures using the same 3D density representation as for docking. Superposition is very much like docking, except now the search is for maximum similarity rather than maximum complementarity. Superposition calculations can only be performed once you have opened a receptor and a ligand molecule. If you want to superpose a pair of molecules that originate from the same PDB file, you will have to manually edit the molecules into separate PDB-format files. Superposition calculations are controlled by the parameters in the Matching Control panel. To calculate a superposition using the default settings (which are usually reasonable), use:
Controls ... Matching ... Activate |
Hex will take a few seconds to calculate the steric density functions for each molecule. It will then systematically search over the range of angular and intermolecular distance samples given in the Matching Control panel. However, unlike docking, Hex first translates the ligand's centre of mass to coincide with that of the receptor. If the molecules are similar, this essentially reduces the search space to a rotational search (to a first approximation). Hence, the default distance range and angular samples are set to much smaller values than are used in a typical docking correlation search. Similarly, good global overlaps can be found using much lower-order correlations than needed for docking. Therefore, its usually sensible to leave all the superposition search parameters at their default values. Nonetheless, the defaults generally give a search which significantly over-samples the search space, and only one or two distinct superpositions are likely to remain after clustering.
By default, superpositions are calculated using only the van der Waals density representation of each molecule. However, this can be changed with the Search Mode selector to match shapes using both the interior (van der Waals) and exterior (surface skin) density functions. This doubles the computational cost, but it can be useful when matching a small molecular fragment onto a surface patch of a larger molecule, because the skin-matching component ensures surface matches are favoured over trivial inclusion matches of the fragment within the body of the larger molecule.
The shape matching algorithm re-uses much of the docking code, and the Matching Control panel works in almost exactly the same way as for docking (see section Docking Examples). One noticeable difference, however, is that similarity scores are calculated using a Carbo-like overlap score. This is essentially a normalised overlap volume, scaled onto [-1000,0] for compatibility with the existing clustering, ranking, and summary output code used by the docking module. In other words, Hex always thinks “negative is good.”
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In Hex, macros (or scripts) are command files which contain a sequence of instructions equivalent to the commands that you might issue from the graphical user interface. To create a macro file, go to the Macros menu and select Learn. All subsequent operations that you perform (including opening files, editing molecules, changing colours, etc.) will be recorded. When you have completed the sequence of commands that you want to record, select the Save command in the Macros menu. This will produce a small window, prompting you for a file name in which to save the command sequence. The convention is to use a file name of the form filename.mac, although you could adopt your own convention.
Here is an example. Lets open an antibody structure, colour it according to the residue identities of the framework and hypervariable loop regions, and position the molecule (using the mouse) in a pleasing orientation. The command sequence might be:
Macros ... Learn # start recording File ... Open ... Receptor ... 3hfl.pdb # existing file Controls ... Molecule ... Colour File ... 3hfl.col # existing file Controls ... Hetero ... Display Solvent ... solvent display off Lock # lock home view Macros ... Save ... test.mac # save commands |
Printing the file test.mac should show a command sequence similar to:
open_receptor 3hfl.pdb set_colour_file 3hfl.col display_solvent 0 button_active 3 cursor_rotation -0.0161 0.0233 button_active 0 button_active 2 cursor_rotation 0.0121 -0.0295 button_active 0 lock_home_view 1 |
You can now re-execute these commands using:
File ... Close ... All Macros ... Run ... test.mac |
If you make a mistake while recording a macro and want to cancel the current recording, select:
Macros ... Forget |
Note that running a macro does not update the state of any toggles or any numerical values in the graphical user interface. Thus the state of the display and the values shown in the GUI controls may become inconsistent after running a macro. Nonetheless, macros are useful for running canned sequences of commands and for running batch jobs. Since a macro file may invoke another macro, it is quite easy to set up a sequence of docking calculations, for example, using the same docking parameters on a number of trial complexes.
Here is a more complicated example using several macro files, starting with the top level macro, dock_ptc.mac, which is provided in the examples directory:
#------------------------------------------------------------------- # # dock_ptc.mac # docking_receptor_samples 492 docking_ligand_samples 492 docking_alpha_samples 128 receptor_range_angle 180 ligand_range_angle 30 twist_range_angle 360 r12_range 21 r12_step 0.75 grid_size 0.6 docking_main_scan 16 docking_main_search 25 run_macro ptc.mac run_macro randomise_ligand.mac activate_docking #------------------------------------------------------------------- # # ptc.mac - loads unbound protein subunits in standard orientation # close_all open_receptor 2ptn_e.pdb open_ligand 4pti_i.pdb open_complex 2ptc.pdb fit_ligand #------------------------------------------------------------------- # # randomise_ligand.mac # moving_thing 1 # (edit 0=receptor, 1=ligand, 2=complex) randomise_molecule # (i.e. randomise the ligand orientation) commit_view # (commit changes) moving_thing -1 # (done editing) #------------------------------------------------------------------- |
One could run this
macro sequence interactively, and then use the
Page/Up and Page/Down keys to step
through displayed solutions. However, when testing the docking algorithm,
or when performing multiple docking runs, its probably easier
(and of more interest) to examine
the calculated energies and ranks of each predicted docking orientation.
Since these values are printed as the calculations proceed,
its often convenient to run the whole thing in batch and to capture
the results in a log file.
For example, at the unix prompt, type:
hex <dock_ptc.mac >dock_ptc.log # Hex goes into the background ps -ef | grep hex # check background process running |
The various docking parameters are described in more detail in Docking Molecules.
Please be aware that
very little checking is done when reading a macro file.
Hence you should always use the GUI,
via Macros ... Learn and Macros ... Save,
to find legal values for the macro commands. Using values which are not
available in the GUI may crash the program or produce meaningless results.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
When Hex loads a protein or DNA structure, it tries to draw all atoms including any water molecules or other hetero atoms (such as ions and other small molecules) that it finds in the PDB file. The Hetero Control panel allows a certain degree of control over how such atoms are treated. The Hetero Control panel is activated using:
Controls ... Hetero |
This panel allows you to modify the treatment of solvent and hetero atoms:
Display Solvent (toggle) Enable Solvent (toggle) Display Hetero (toggle) Enable Hetero (toggle) |
When Enable Solvent is selected, all water molecules are treated as part of the structure when calculating dot surfaces or spherical harmonic surfaces, for example. Toggling this option excludes solvent from the surface calculation. The Display Solvent toggle may be used to eliminate all waters from the graphics display (but remember, they still exist (if enabled), as far as any calculations are concerned). Enabled solvent atoms are displayed in light blue: disabled atoms are displayed in grey. The Hetero toggles have a similar function. Similarly, the Enable ARG/LYS toggle allows the status of any arginine and lysine side chain atoms to be changed. Disabling these side chains during a predictive docking run generally reduces the sensitivity of the docking correlation, but is often highly beneficial in the following molecular mechanics refinement step: This part of the program is still under development.
The Solvent Colour button activates a simple colour chart which may be used to select an alternative colour with which to display enabled solvent atoms.
The Cull Solvent button calculates which water molecules are completely buried within a protein - and disables those that are not. The culling calculation uses the dot surface algorithm to determine the accessibility of each water molecule. The van der Waals surface of each atom in the structure is covered with dots: those dots which are occluded by neighbouring atoms are deleted in a recursive search, so that any atom with no surviving dots must be completely inaccessible to a probe sphere. Any water molecule that has surviving surface dots must, therefore, be accessible to the probe and is culled: i.e. removed from the surface atom list. The algorithm is repeated (automatically) until no more waters can be removed. Usually this takes from one to four cycles, depending on how many shells of solvent surround the molecule.
As an alternative to this automatic calculation, it is also possible to manually select water molecules (or any other atoms) to be culled. Select ID picking mode, and pick those atoms you wish to cull. Pressing the Toggle Picked IDs button (in Hetero Control) will toggle the status of any picked atoms.
Finally, using the Apply to selector, you can choose to apply these operations to all molecules (the default) or only to a selected molecule. If more sophisticated atom selections are required, it will probably be necessary to edit PDB files manually, before loading them into the program.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex supports JPEG, PNG, and Postscript hardcopy output.
Just compose your scene
and then use the appropriate File ... Print option to write the
scene to an image file. The File ... Print menu also contains a
special Pipe option. This causes the molecular coordinates for the
current scene to be piped (in the sense of a Unix pipe) to a script in
the $HEX_ROOT/pipes directory, which can be used to load the
current structures directly into an external program such as GRASP or
Molscript. The $HEX_ROOT/pipes directory contains example scripts
to run these programs.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex has full support for gzip (.gz) and bzip2 (.bz/.bz2) file compression formats. If you specify an output file name with one of these file name extensions, you will automatically get the corresponding type of compressed output. Similarly, if any input file has one of these extensions, Hex will automatically decompress the file as it reads it. If you have installed a copy of the PDB (e.g. from the PDB CDROM disc set) on your hard drive, you can use Hex to view the structure files without first having to decompress them. Just point Hex's file selection widget at the top level of your PDB directory and start visual browsing!
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex uses several environment variables to locate directories and data files. Most of these are optional, as described in the table below. The non-optional environment variables are defined automatically if you use the self-installing bundles or the hex_configure.bin script to install Hex.
Name | Status | Default | Description |
HEX_CACHE | optional | $HEX_ROOT/cache | locates a cache directory for the translation matrices: highly recommended |
HEX_COLOURS | optional | $HEX_ROOT/examples | locates a directory of Hex colour files |
HEX_CPUS | optional | 1 | the default number of CPUs to use |
HEX_GPUS | optional | 1 | the default number of GPUs to use |
HEX_FIRST_GPU | optional | 0 | defines the first GPU to use for computations |
HEX_DATA | optional | $HEX_ROOT/data | locates non-standard Hex data files |
HEX_LOG | optional | hex.log | defines the name of the current log file |
HEX_MACROS | optional | $HEX_ROOT/examples | locates a directory of Hex macro files |
HEX_PDB | optional | $HEX_ROOT/examples | locates a directory of PDB files |
HEX_PIPE | optional | $HEX_ROOT/pipes | locates a directory of Hex pipe scripts |
HEX_MESSAGES | optional | yes | write messages to separate output window |
HEX_ROOT | required | none | locates the main Hex installation directory (e.g. /usr/local/hex) |
HEX_STARTUP | optional | none | locates a start-up macro script (e.g. $HEX_ROOT/data/startup_v4.mac) |
HEX_STEREO | optional | yes | defines whether a stereographic visual is requested by default |
HEX_VERSION | optional | 6a | the default version of Hex to use |
Additionally, you should ensure that your PATH contains the $HEX_ROOT/bin directory. On systems with multiple GPUs, it may be desirable to reserve some GPUs for display or other compute purposes. For example on a system with 3 GPUs, you could force Hex to use only the last GPU by setting HEX_FIRST_GPU=2 and HEX_GPUS=1.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The general command-line format to invoke Hex is
hex [-options] [pdbfile(s)] |
where options may be a combination of:
-v# (where # is a version number/string - e.g. 4f, 5a) -ncpu n (where n is the number of CPUs to use) -ngpu n (where n is the number of GPUs to use) -nice n (runs at lower priority: n a "nice" value) -cuda (request program with CUDA support; the default) -nocuda (request program with no CUDA support) -kill (try to kill any stuck Hex jobs) -g (run within a debugger such as gdb) -debug (print debugging messages; another -debug adds more) -noexec (don't put a batch run in the background) -gui (force GUI mode even when stdin is not a terminal) -stereo (request a stereographic window) -s (same as -stereo) -nostereo (request a non-stereographic window) -nos (same as -nostereo) -messages (write messages to a separate window - the default) -nomessages (write messages to standard output, not separate window) -stdout (same as -nomessages) -log logfile (or -l, write log of entire session to file) -l logfile (same as -log) -pdb pdbfiles (load up to 3 PDB files to be displayed) -browse (same as -pdb, but forces interactive mode for browser) -noligand (treat two PDB files as receptor + complex) -2 (short form of -noligand: superposes two molecules) -pos (position ligand near receptor - the default) -nopos (don't mess with ligand coordinates) -e macfile(s) (execute up to 3 macro files) |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Load three PDB files which contain a receptor, a ligand, and an existing complex structure (to be used to calculate RMS deviations of the docking predictions), and execute a macro file of docking commands, writing the results to a named log file:
hex r.pdb l.pdb c.pdb -e docking_job.mac -l docking_job.log |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Load a receptor and a complex (i.e. no ligand), and superpose the complex onto the receptor:
hex -2 r.pdb c.pdb |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Run a docking calculation (assumed to be fully specified in a macro file), using 2 CPUs:
hex -ncpu 2 -e job.mac -l job.log |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Run the previous example using I/O redirection:
hex -ncpu 2 <job.mac >job.log |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
If both stdin and stdout are not terminal devices,
Hex automatically puts itself into the background. When running a Unix
script with a series of different jobs, this behaviour should be suppressed
using the -noexec option:
hex -ncpu 2 -noexec <job1.mac >job1.log # job2 starts after job1 hex -ncpu 2 -noexec <job2.mac >job2.log |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
On Unix systems, Hex may be used as a web browser helper application for PDB files by specifying that Hex should be invoked when the browser receives a MIME type that corresponds to a PDB file (e.g. chemical/x-pdb). See the hex_browser file in the $HEX_ROOT/bin directory for details. If anyone implements something similar for MS-Windows, please let me know!
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The examples directory, under the main hex directory, contains PDB data files of examples of two protein complexes: HyHel-5/lysozyme and BPTI/Inhibitor. It also contains short macro files to load the subunits of these complexes. Unless you have changed the values of the HEX_PDB and HEX_MACROS environment variables, Hex will look in the examples directory by default. Select:
Macros ... Run |
and then select one of [3hfl.mac, 2ptc.mac], or [hfl.mac, ptc.mac]. This will open the files that contain the atom coordinates of either the crystallographically determined complex (first pair) or the coordinates of the separately determined protein subunits (second pair), superposed on the corresponding complex structure. In both cases the structure of the complex should appear as a thin grey skeleton.
The examples directory also contains sample colour files, *.col, and a few other macro files, *.mac, which you might wish to examine. In particular, the startup.mac file explains how to select an initial colour scheme, for example. It also shows how to switch off the initial logo display if you prefer to start with a blank screen, and how to change the type of browser (Netscape, by default) used to display this User Manual on-line.
Please note, the example docking scripts dock*.mac will only work correctly if Hex can find the relevant PDB files and related macros. Once you've defined the HEX_PDB and HEX_MACRO environment variables, to point to your own directories, you should copy the appropriate files from the examples directory into these new directories if you still want to use these scripts. Optionally, you can also download a more extensive set of docking examples from the Hex download page. A much more extensive set of docking test cases can be obtained from Zhiping Weng group's Docking Benchmark.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Please report any bugs/suggestions to the author (see section Contacts). Described below are some known limitations, some of which might never be fixed.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
When Hex runs a macro, it doesn't update the state of any toggles or any numerical values in the graphical user interface. Thus the state of the display and the values shown in the user interface controls may become inconsistent after running a macro.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Non-standard or substituted amino acid residues may not be drawn properly,
because Hex requires an entry in the atom_type.dat
and atom_bonds.dat files for each type of atom and bond, respectively.
However, Hex makes a reasonable attempt to draw the bonds of
unrecognised molecules (typically small ligands).
The sequence File ... Open ... Dataset is intended to allow
user-specific atom and bond data files to be loaded, but this isn't properly
implemented yet.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The Graphics Font selected from the font menu panel is not propagated to Postscript. All printed text always appears in Times-Roman.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Surface sampling using an icosahedral tessellation and surface parametrisation using spherical harmonic functions are both central to the shape similarity and complementarity calculations in Hex. In Test Mode, you can view the spherical harmonic basis functions and the icosahedral tessellations used in the calculations. Go to the File menu and select:
File ... Open ... Test |
You now should see a spherical grid plotted as a white mesh. Each grid cell has approximately the same area. Formerly, this grid was used as a look-up table to map an arbitrary spherical coordinate (theta,phi) onto the nearest vertex of an icosahedral tessellation, but its no longer used in the current version. You can change the number of grid cells using the Mesh Order slider in the Harmonic Surface control panel. Select:
Graphics ... Harmonic Surface ... Mesh Order ... 10 |
The Mesh Order slider controls the number of vertices to be used in an icosahedral tessellation, by specifying the number of subdivisions made along each edge of a regular icosahedron. (It also indirectly controls the number of cells in the corresponding spherical grid).
You can view the icosahedral tessellation by selecting the L=0 spherical harmonic function:
Controls ... Harmonics ... Order ... 0 |
and by clicking the Enable Harmonics button. You may need to adjust the zoom factor (bottom left slider) to get a better view. The Harmonics button (which looks like a balloon: its supposed to represent the L=2, M=0 harmonic function) on the right-hand border of the main window also acts as a toggle equivalent to the Enable Harmonics button.
You can now display arbitrary spherical harmonic functions using the Order (L) and Degree (M) sliders. Each function is plotted by evaluating the function at the angular coordinates of each icosahedral tessellation vertex and by setting the radial distance for each angular sample to that of the function value. You can control the number of sample points with the Mesh Order slider (above) and you can control the display style of the functions using the Display Mode selector of the Harmonic Surface control panel. Try:
Graphics ... Harmonic Surface ... Display Mode ... Polyframe |
Note that when you rotate or translate the scene using the mouse buttons, the display reverts to wire-frame rendering until you release the button. This is usually faster than re-rendering polygons during the motion. The Solid Motion button (right-hand border, last button) can be used to toggle this behaviour.
The three sliders in the Harmonics Control panel labelled Alpha, Beta and Gamma are used to test Euler angle rotations of the spherical harmonics. Changing these values will cause the rotated function R(alpha,beta,gamma)(Y[l,m](theta,phi)) to be calculated and displayed. Visually, this should be identical to drawing the unrotated figure and then manually rotating it with the mouse by R(gamma) about the z-axis, R(beta) about the y-axis, followed by R(alpha) about the z-axis. This is an important visual test because ensuring that the spherical harmonic functions can be rotated computationally is fundamental to Hex's shape superposition and docking calculations.
Finally, using spherical harmonics to parametrise the surface shape of a cube provides a further (possibly perverse) test of the method. You could try:
File ... Open ... cube |
and then experiment with different display styles and surface resolutions. Using L=16 gives a remarkably good spherical harmonic cube. In addition to rotating the cube manually, it can be rotated by rotating the harmonic basis functions (above) or, equivalently, by rotating the shape expansion coefficients by setting the Rotate Coefficients toggle in Harmonics Control.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In order to run Hex, your system should have at least 64Mb main memory but preferably more (128 Mb or greater), and about 4 MB of disc space for the program and data files. The speed of the docking calculations can be increased by allocating an optional disc cache directory of up to about 100 MB. For molecular docking and graphics applications, I'd recommend buying the highest-spec Linux machine you can afford, but be sure to select a graphics card that has Linux support. I personally own a Pentium III Xeon system with a Quadro4 XGL700 graphics card (currently running Redhat 9, and Fedora 4 and 8). Its great.
The latest Linux version was built on Pentium III Xeon system running Fedora Core 8 (kernel 2.6.23, X.org 7.1, gcc 4.1). Versions are also available for Fedora Core 4 (kernel 2.6.21, X.org 6.8.2, gcc 4.0), RedHat 9.0 (kernel 2.4.20-8, XFree86 4.3.0, gcc 3.2.2), and RedHat 8.0 (kernel 2.4.18-14, XFree86, 4.2.0, gcc 3.2-7). Earlier versions of Hex are still available for some older versions of RedHat.
As of Hex 4.5, PowerPC executables are now available for Mac OS X (Tiger 10.4), and Linux (Fedora Core 8). These were built on a G4 PowerBook, but should work on other PowerPC systems. An version for the Intel Mac will be made available soon.
The Windows version was built on a Pentium III 686 system running Windows XP, and using the MinGW development environment. As far as I know, the Windows executable works on all recent Windows systems.
Due to lack of available hardware and rapidly falling demand, executables for SGI/IRIX and Sun/sparc are now only available up to version 4.5. Sorry.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Author: Dave Ritchie. Before e-mailing me with a problem or question, please first check the FAQs (see section Frequently Asked Questions). I always try to respond to problems, and I welcome suggestions for future enhancements. However, if you do have a question, suggestion, or if you think you've found a bug, please check the FAQs first (see section Frequently Asked Questions), and check Hex's Home Page to make sure you're using the lastest version. Please remember that Hex is essentially a research program and it is provided “as is”, without any guarantee of updates or user-support.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Much of Hex was developed during projects (1996-2000) funded by the BBSRC. Hex was written mostly in C, but also uses some C++ for the GUI. The graphical user interface was built with FLTK. The Hex logo and icons were created with The Gimp. The translation matrices used in Hex are calculated using the GMP library. The 3D and 5D FFTs were implemented on Linux using the Intel MKL library, and on other platforms using Kiss FFT, Copyright (c) 2003-2004, Mark Borgerding. Support for compressed files is thanks to zlib, gzip, and bzip2. The initial Linux version of Hex would have been impossible without the Mesa graphics library. The Windows version of Hex was built entirely under the MinGW environment (Hex has never been anywhere near a Microsoft compiler). This manual was produced using texinfo.
Hex uses the Stride program to assign protein secondary structures for cartoon displays. See section Stride Licence if you have any doubts about whether you are entitled to use Stride.
Vishwesh Venkatraman helped to re-write the CPU and GPU correlation calculations to use multi-threading.
The following people for making significant programming contributions to Hex as part of their Honours or MSc projects: Ryan Cairns BSc (FLTK port), Ewan Lyle BSc (ribbon cartoons), Ioannis Zaferopoulos BSc (solid models), Russell Hamilton MSc (macro docking displays), Guillaume Valadon BSc (surface colouring).
Gary MacIndoe wrote a nice web interface: http://www.loria.fr/~ritchied/hex_server/. This is useful if you wish to try out Hex without having to install it. However, for best results, please download the program from Hex, and run it locally.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
When citing Hex in a publication, please cite whichever of the above articles seems most appropriate, and please also mention Hex's Home Page:
http://www.loria.fr/~ritchied/hex/ |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
This is a legal agreement between you, the LICENSEE, and the LICENSOR for use of the computer program Hex 6.X, where "6.X" is a version number. This licence does not apply to versions of Hex with version numbers less than "6.0".
By installing copying or otherwise using Hex 6.X you agree to be bound by the terms of this licence agreement. If you do not agree to the terms of this licence agreement the licensor is unwilling to license the software to you. In such circumstances, you may not use or copy the software, and you should return the unused product within 7 days.
1. Definitions
(a) LICENCE means the terms and conditions contained herein.
(b) LICENSOR means
the Institut National de Recherche en Informatique et en Automatique (INRIA),
of Domaine de Voluceau, Rocquencourt, Le Chesnay, France,
as owner of the software comprising the computer program Hex 6.X.
(c) SITE means a single
computer or internal computer network operated by the LICENSEE at the premises
of the LICENSEE specified when Hex 6.X was ordered or installed.
2. Grant of Licence
(a) The LICENSOR grants
the LICENSEE a non-transferable and non-exclusive licence to use Hex 6.X
in connection with computers only on the SITE to which the LICENCE applies.
(b) The LICENSEE will
not, under any circumstances, reverse engineer, decompile or disassemble
Hex 6.X.
3. Licence Fee
(a) In consideration
of the LICENCE granted pursuant to Section 2, the LICENSEE shall pay to
the LICENSOR the applicable LICENCE FEE within 30 days of receipt of an
appropriate invoice prepared by the LICENSOR.
(b) For Academic and
Governmental SITES, the LICENCE FEE is waived.
4. Transfer of Licence
(a) The LICENSEE shall not in any way sublicense, rent, assign or transfer this LICENCE or the computer program Hex 6.X.
5. Copyright
(a) The LICENSOR owns
the copyright and all and any other intellectual and industrial property
rights in Hex 6.X and its associated documentation. You must therefore treat
Hex 6.X and its associated documentation like any other copyrighted material
(e.g. a book).
(b) Hex 6.X and the
associated documentation is protected by United Kingdom copyright laws,
international treaty provisions, and all other applicable national laws.
(c) The LICENSEE acknowledges
that it does not and shall not acquire any title, copyright or any other
intellectual property rights in Hex 6.X or the associated documentation.
6. Limited Warranty and Disclaimer of Warranty
(a) The LICENSEE acknowledges
that software in general is not error-free and that the existence of such
errors shall not constitute a breach of this LICENCE.
(b) Save as expressly
provided under this LICENCE, no warranty or conditions express or implied,
statutory or otherwise as to condition, quality, performance, merchantability
or fitness for purpose is given or assumed by the LICENSOR in respect of
Hex 6.X and
its associated documentation and all such warranties and conditions
are hereby excluded save to the extent that this exclusion is prohibited
by law.
7. Limitations of Liability
(a) Except to the extent
that such liability may not lawfully be excluded, the LICENSOR shall not
be liable for damages arising out of or in conjunction with the LICENCE
of Hex 6.X or its use or performance.
(b) In no event shall
the LICENSOR be liable for any loss of profits, business interruption,
loss of business information or other special consequential damages, even
if the LICENSOR has been advised of the possibility of such damages, or
for any claim against the LICENSEE by any other party.
8. Termination
(a) This LICENCE agreement
is effective from the date of purchase or installation, whichever is earlier,
of the computer program Hex 6.X.
(b) The LICENSEE can
terminate the LICENCE at any time by notifying the LICENSOR of its intention
in writing.
(c) The LICENSOR can
terminate the LICENCE, after providing the LICENSEE with 14 days prior
notice and an opportunity to remedy the situation, if the LICENSEE fails
to pay any amount due to the LICENSOR.
(d) The LICENCE shall
terminate 30 days after written notice by either party in the event of
any material or persistent breach by the other party of any of the terms
of this LICENCE which is either incapable of remedy or remains unremedied
at the end of the said period of 30 days.
(e) Upon termination
of this LICENCE, the LICENSEE shall (i) discontinue use of Hex 6.X (ii) deliver
to the LICENSOR all the software furnished by the LICENSEE pursuant hereto
together with all copies thereof and (iii) erase or destroy any of Hex 6.X
contained in the computer memory or data storage systems under the control
of the LICENSEE.
9. Entire Agreement
(a) This agreement constitutes the complete and exclusive statement of the agreement between the LICENSEE and the LICENSOR and supercedes all other oral or written proposals, prior agreements and other prior communications between the parties, concerning the subject matter of this agreement.
10. General
(a) This agreement
shall be governed by and construed in accordance with the laws of France,
and the parties prorogate the exclusive jurisdiction of the French Courts.
(b) No representation
or promise relating to, and no amendment of, this agreement shall be binding
unless it is in writing and signed by both parties.
(c) The terms and
conditions of this agreement shall previal notwithstanding any variance
with the terms and conditions of any order submitted by the LICENSEE.
(d) Except for the
LICENSEE's obligation to pay the LICENSOR, neither party shall be liable
for any failure to perform due to cause beyond reasonable control.
(e) No waiver by a
party of any breach of any provision of this agreement shall constitute
a waiver of any other breach of that or any other provision of this agreement.
(f) In the event that
any of the provisions of this LICENCE are held to be invalid or unenforceable,
this agreement shall be construed without such provisions.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The following text is reproduced from the Stride Documentation:
“All rights reserved, whether the whole or part of the program is concerned. Permission to use, copy and modify this software and its documentation is granted for academic use provided that:
(i) this copyright notice appears in all copies of the software and related documentation;
(ii) the reference given below (Frishman and Argos, 1995) must be cited in any publication of scientific results based in part or completely on the use of this program;
(iii) bugs will be reported to the authors.
The use of the software in commercial activities is not allowed without a prior written commercial license agreement."
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
The following text is reproduced from the Kiss FFT source code:
“Copyright (c) 2003-2004, Mark Borgerding
All rights reserved.
Redistribution and use in source and binary forms, with or without modification, are permitted provided that the following conditions are met:
* Redistributions of source code must retain the above copyright notice, this list of conditions and the following disclaimer.
* Redistributions in binary form must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution.
* Neither the author nor the names of any contributors may be used to endorse or promote products derived from this software without specific prior written permission.
THIS SOFTWARE IS PROVIDED BY THE COPYRIGHT HOLDERS AND CONTRIBUTORS "AS IS" AND ANY EXPRESS OR IMPLIED WARRANTIES, INCLUDING, BUT NOT LIMITED TO, THE IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE ARE DISCLAIMED. IN NO EVENT SHALL THE COPYRIGHT OWNER OR CONTRIBUTORS BE LIABLE FOR ANY DIRECT, INDIRECT, INCIDENTAL, SPECIAL, EXEMPLARY, OR CONSEQUENTIAL DAMAGES (INCLUDING, BUT NOT LIMITED TO, PROCUREMENT OF SUBSTITUTE GOODS OR SERVICES; LOSS OF USE, DATA, OR PROFITS; OR BUSINESS INTERRUPTION) HOWEVER CAUSED AND ON ANY THEORY OF LIABILITY, WHETHER IN CONTRACT, STRICT LIABILITY, OR TORT (INCLUDING NEGLIGENCE OR OTHERWISE) ARISING IN ANY WAY OUT OF THE USE OF THIS SOFTWARE, EVEN IF ADVISED OF THE POSSIBILITY OF SUCH DAMAGE.”
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
For Microsoft Windows, just download and run the self-installer program hex-6a-win.exe. This adds a Hex icon to the Programs menu and adds the environment variable HEX_ROOT to the registry. That's all you should need to get started! However, there are several other environment variables which you can set to customise Hex's behaviour. These have the same names as the Linux version described below. In particular you may wish to define the HEX_STEREO and HEX_CACHE variables.
By default, Hex will use a stereographics window if a suitable graphics card is detected. However, this gives a slower drawing rate than non-stereo mode, even when viewing a non-stereo (zero parallax) scene. Setting HEX_STEREO=no causes a non-stereographic window to be selected at start-up time, and hence should give faster graphics performance. During a docking calculation, Hex will write translation matrix files to the $HEX_ROOT\cache directory. However, you can define HEX_CACHE to point to another directory (possibly on another disc with more space) to over-ride the default.
In Windows XP, environment variables are defined using the
Advanced option in the System control panel.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
For Mac OS X, just download and double-click the
hex-6a-osx.pkg package file. This will install Hex as
an application bundle in the /Applications directory. Double-clicking
the Hex icon will run the program. The bundle automatically assigns
the correct value of the HEX_ROOT environment variable.
You may wish to edit the hex_launch script within the bundle to
assign non-default values to the other environment variables mentioned
in this section.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
You will need to download the installation script hex_setup.bin, and either the hex-6a-lnx.tgz tar file for an Intel PC or hex-6a-ppc.tgz for Linux on a PowerPC machine (i.e. not running Mac OS X). The distribution files should be installed using the shell script hex_setup.bin by typing:
sh hex_setup.bin |
This script can normally be trusted to install Hex correctly,
even if you
have already installed an earlier version of the program. The script
will add any necessary environment variables to your default shell
environment.
In cases where a central file server is used to serve files to different
types of workstations, several different executables may be installed into
a single hex installation directory.
Note that in previous versions of Hex, the installation script
was called hex_install.bin.
If you prefer to install the program manually, you should
create a hex directory, cd to that directory,
and then uncompress the distribution files into it.
For example, assuming you are installing the usual Linux PC version, use:
zcat hex-6a-lnx.tgz | tar xvpf - |
In order to make Hex work properly, your login script (.login or .profile, or whatever) should set the environment variable HEX_ROOT to the hex installation directory. You should also add Hex's bin directory to your PATH so that (a) you can run Hex, and (b) Hex can find the files it needs under HEX_ROOT. These two steps are normally performed by the hex_setup.bin script. Optionally, you can also specify directories that specify where Hex should start looking for colour files, macro files, and PDB files, and you can specify the location of a temporary cache directory (all except the hex installation directories are optional and must be created manually). You can also specify the default number of CPUs to use on a multi-processor system. These parameters are described in Environment Variables.
Here's a C-shell example (I have these lines in my .cshrc file):
setenv HEX_ROOT /home/ritchied/hex
setenv HEX_PDB .
setenv HEX_MACROS .
setenv HEX_COLOUR .
setenv HEX_DATA /home/ritchied/hex/data
setenv HEX_CACHE /tmp/ritchied/hex_cache
setenv HEX_STARTUP /home/ritchied/hex/examples/my_startup_v5.mac
setenv HEX_CPUS 8
setenv HEX_GPUS 2
setenv HEX_MESSAGES yes
setenv HEX_STEREO yes
setenv PATH ${PATH}:${HEX_ROOT}/bin
|
For Bourne and Korn-shell users, the corresponding commands (in the .profile file) would be:
set HEX_ROOT = /home/ritchied/hex
set HEX_PDB = .
set HEX_MACROS = .
set HEX_COLOUR = .
set HEX_DATA = /home/ritchied/hex/data
set HEX_CACHE = /tmp/ritchied/hex_cache
set HEX_STARTUP = /home/ritchied/hex/examples/my_startup_v5.mac
set HEX_CPUS = 8
set HEX_GPUS = 2
set HEX_MESSAGES = yes
set HEX_STEREO = yes
set PATH = ${PATH}:${HEX_ROOT}/bin
export PATH HEX_CACHE HEX_STARTUP HEX_CPUS HEX_STEREO
export HEX_ROOT HEX_PDB HEX_MACROS HEX_COLOUR HEX_DATA
|
In addition to the README and LICENCE files, the hex directory should contain the following 7 directories:
bin exe doc data examples pipes cache |
Typing hex at the Linux command prompt should locate the
script hex
in the bin directory which
then launches the appropriate executable from the exe directory.
As of version 6.0, there are normally two separate executables for each type of
system: one build with CUDA and one built without CUDA. By default, the launch
script will run the CUDA executable if it has been installed. You can give the
option -nocuda on the command line to select explicitly the non-CUDA executable.
The examples directory contains several PDB files and example Hex macro files. Unless you set the above environment variables, Hex will first look in the examples directory for any macro and PDB files you want to use. The data directory contains the runtime data files necessary to colour and charge protein and DNA molecules, for example. The pipes directory contains Linux scripts which allow the molecular structures in the current Hex scene to be written directly to external programs such as GRASP or Molscript. The cache directory is the default location in which Hex will store its translation matrix files for docking runs. You may wish to use a different location for the cache by defining the HEX_CACHE environment variable to point to an alternative directory.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex has been on-going work for around 12 years, although most of the basic functionality was implemented in the first three years. This section summarises the development history. There are no entries here for versions 4.3 and 4.4 - these were internal development versions and were never released.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex Version 4.5.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex Version 4.2.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex 4.1 is a bug-fix release.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex 3.3 is a bug-fix release. Running Hex in batch mode (or when standard input was not a terminal device) was broken. The program would quickly crash with a SIGSEG error. Fixed now.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex 3.2 contains mostly minor bugfixes. However, stereo graphics is now available on Linux systems that have suitable hardware support:
hex -stereo
to request stereo (see section Stereo Displays).
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex 3.1 represents a fairly major upgrade relative to Hex 3.0, as follows:
F to toggle (see section Full Screen Mode).
hex -kill to clean up stuck
processes (see section Command Line Options).
Note, there is still technically a PDB limit of 99,999 atoms per structure (Fortran I5 format), but Hex will try to read files that break this restriction. As of version 3.1, the models in multi-model structures are now numbered from 1, or use the actual model number given in the PDB MODEL record.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Hex 3.0 contains several new features and a number of improvements compared to Hex 2.4. These are mostly concerned with the docking algorithm and the graphics displays, although several bugs reported by users have also been fixed. The main new features are:
Existing Hex 2.4 users: Please note that some of the parameter names used in macro files have changed from version 2.4. Therefore, some of your existing macro files may no longer work as expected. This was unavoidable, given the many changes in the new version. Apologies for any inconvenience.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
In order to use one or more Nvidia GPU devices in a docking calculation, it is necessary to download a version of Hex wich was compiled with CUDA support, and to install a CUDA-enabled device driver and the cuFFT run-time library, which is part of the CUDA developer Toolkit (http://www.nvidia.com/object/cuda_get.html, Get CUDA).
On Windows systems, Hex plus CUDA should "just work". Some Linux systems (e.g. Ubuntu) provide a convenient way to download the Nvidia driver as a "proprietary hardware driver". However, I recommend that you do NOT use this feature as it could cause problems when downloading the developer Toolkit. I recommend downloading both driver and toolkit directly from the Nvidia web site. However, if you upgrade the Linux kernel, you will need to re-install the Nvidia drivers.
In order to let the Hex executable find the CUDA run-time libraries, you will need either to set the LD_LIBRARY_PATH environment variable to point to the folder containing the CUDA libraries, or you will need to edit the system file /etc/ld.so.conf to mention the location of the CUDA libraries if they were not in a well-known place such as /usr/lib. On my 64-bit workstation, I installed CUDA in /usr/local/cuda. I then edited /etc/ld.so.conf to make it look like:
include /etc/ld.so.conf.d/*.conf # # make CUDA run-time stuff globally visible # /usr/local/cuda/lib64 |
The /usr/bin/ldd tool make be used to check that all run-time libraries are visible to the Hex executable. Here is the result on my system:
jordan$ ldd /home/ritchied/hex/exe/hex6-cuda.x64 linux-vdso.so.1 => (0x00007fff013bf000) libpthread.so.0 => /lib/libpthread.so.0 (0x00007f6b30e49000) libcufft.so.2 => /usr/local/cuda/lib64/libcufft.so.2 (0x00007f6b303f4000) libcudart.so.2 => /usr/local/cuda/lib64/libcudart.so.2 (0x00007f6b301b4000) libcuda.so.1 => /usr/lib/libcuda.so.1 (0x00007f6b2c532000) libGLU.so.1 => /usr/lib/libGLU.so.1 (0x00007f6b2c2c1000) libGL.so.1 => /usr/lib/libGL.so.1 (0x00007f6b2c0c2000) libXft.so.2 => /usr/lib/libXft.so.2 (0x00007f6b2bead000) libXrender.so.1 => /usr/lib/libXrender.so.1 (0x00007f6b2bca3000) libfontconfig.so.1 => /usr/lib/libfontconfig.so.1 (0x00007f6b2ba71000) libexpat.so.1 => /lib/libexpat.so.1 (0x00007f6b2b848000) libfreetype.so.6 => /usr/lib/libfreetype.so.6 (0x00007f6b2b5c3000) libXext.so.6 => /usr/lib/libXext.so.6 (0x00007f6b2b3b1000) libX11.so.6 => /usr/lib/libX11.so.6 (0x00007f6b2b07b000) libm.so.6 => /lib/libm.so.6 (0x00007f6b2adf7000) libgcc_s.so.1 => /lib/libgcc_s.so.1 (0x00007f6b2abe0000) libc.so.6 => /lib/libc.so.6 (0x00007f6b2a871000) /lib64/ld-linux-x86-64.so.2 (0x00007f6b31065000) libdl.so.2 => /lib/libdl.so.2 (0x00007f6b2a66d000) libstdc++.so.6 => /usr/lib/libstdc++.so.6 (0x00007f6b2a35d000) librt.so.1 => /lib/librt.so.1 (0x00007f6b2a155000) libz.so.1 => /lib/libz.so.1 (0x00007f6b29f3e000) libGLcore.so.1 => /usr/lib/libGLcore.so.1 (0x00007f6b282e7000) libnvidia-tls.so.1 => /usr/lib/tls/libnvidia-tls.so.1 (0x00007f6b3115b000) libXau.so.6 => /usr/lib/libXau.so.6 (0x00007f6b280e4000) libxcb.so.1 => /usr/lib/libxcb.so.1 (0x00007f6b27ec8000) libXdmcp.so.6 => /usr/lib/libXdmcp.so.6 (0x00007f6b27cc3000) |
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
For OpenGL stereo-in-a-window and full-screen stereo, you will need a Quadro FX-1800 graphics card or higher (http://www.nvidia.com/object/quadro_pro_graphics_boards.html, Nvidia Quadro stereo). For stereo on a flat panel display, you will need a device with a refresh rate of 100Hz or higher. Nvidia recommend the Samsung SyncMaster 2233RZ or the Viewsonic VX2265 (http://www.nvidia.com/object/3D_Vision_Requirements.html, flat panel stereo). If you have the right hardware and Windows Vista or Windows 7, stereographics should "just work".
On Linux, configuring your nVidia graphics card for stereo should be straight-forward. Just download the latest driver from http://www.nvidia.com/view.asp?PAGE=linux, and follow the instructions. However, as mentioned above, make sure to de-install any earlier nVidia drivers first.
Here's what the Device Section of my /etc/X11/xorg.conf file looks like after editing nVidia's supplied template:
#
# Stereo is not compatible with the compositing feature:
#
Section "Extensions"
Option "Composite" "Disable"
EndSection
#
# If you have multiple graphics cards, use /usr/bin/lspci to find
# the PCI bus ID of the specific device to use
#
Section "Device"
Identifier "NV AGP"
VendorName "nvidia"
Driver "nvidia"
BusID "PCI:1:0:0"
# Option "Stereo" "0" # 0=off
Option "Stereo" "1" # 1=DCC with cable splitter
# Option "Stereo" "3" # 3=3-pin DIN connector
# Option "Stereo" "10" # 10=USB IR emitter for 3D Vision kit.
Option "metamodes" "DFP: 1680x1050_120 +0+0; DFP: 1680x1050 +0+0"
Option "NoLogo" "true"
Option "Overlay" "false"
Option "UseEdidFreqs" "true"
Option "NvAgp" "1" # 0=off 1=nvidia, 2=AGPGART, 3=try 2, 1
EndSection
|
I verified these settings (and that stereo was enabled) by running the glxinfo program. Although my old XGL700 has an on-board stereo DIN (3-pin) socket, I couldn't get a signal off this with the 3123/4191 drivers. Currently, stereo synching only seems to work in DCC (Data Control Channel) mode. This requires a monitor cable “splitter”, to which the stereo glasses are connected. Such “splitters” often come included with stereo glasses kits. For best performance, use nVidia's AGP driver (part of NVidia's driver distribution), and NOT the Linux AGP driver (/dev/agpgart, etc.). Also, use a kernel built with the MTRR option (memory type-range registers) if your machine supports this.
Provided you have suitable hardware, stereographics on flat panel displays using Nvidia's recent 3D Vision kit should work directly on Windows Vista and Windows 7. For Linux, you will need to download and install a recent version (195.36.15 = 19 march 2010, or later) of the driver (Nvidia drivers, and set Option "Stereo" "10" in the /etc/X11/xorg.conf file. In order to let the nvidia driver detect the USB infra-red emitter, it is necessary to map USB devices to a pseudo-file system (which is not done automatically in Ubuntu Linux). Here is an extract from my /etc/fstab file:
# /etc/fstab: static file system information. # # mount the usb device pseudo file system # none /proc/bus/usb usbfs defaults 0 0 |
Here is a reduced extract from /var/log/Xorg.0.log on my 64-bit FX-5800 workstation.
(**) NVIDIA(0): Option "Stereo" "10" (**) NVIDIA(0): Option "TwinView" "0" (**) NVIDIA(0): Option "MetaModes" "1680x1050_120 +0+0; 1680x1050 +0+0" ... (**) NVIDIA(0): USB IR emitter stereo requested (**) NVIDIA(0): Enabling RENDER acceleration (II) NVIDIA(0): NVIDIA GPU Quadro FX 5800 (GT200GL) at PCI:2:0:0 (GPU-0) (--) NVIDIA(0): Memory: 4194304 kBytes (--) NVIDIA(0): VideoBIOS: 62.00.3a.00.03 (II) NVIDIA(0): Detected PCI Express Link width: 16X (--) NVIDIA(0): Interlaced video modes are supported on this GPU (--) NVIDIA(0): Connected display device(s) on Quadro FX 5800 at PCI:2:0:0: (--) NVIDIA(0): Samsung SyncMaster (DFP-1) (--) NVIDIA(0): Samsung SyncMaster (DFP-1): 330.0 MHz maximum pixel clock (--) NVIDIA(0): Samsung SyncMaster (DFP-1): Internal Dual Link TMDS (II) NVIDIA(0): Validated modes: (II) NVIDIA(0): "DFP:1680x1050_120+0+0" (II) NVIDIA(0): "DFP:1680x1050+0+0" (II) NVIDIA(0): Virtual screen size determined to be 1680 x 1050 (--) NVIDIA(0): DPI set to (88, 88); computed from "UseEdidDpi" X config (--) NVIDIA(0): option (==) NVIDIA(0): Disabling 32-bit ARGB GLX visuals. ... (II) NVIDIA(0): USB IR emitter - Copyright (c) 2009 NVIDIA Corp (II) NVIDIA(0): stereo controller (II) NVIDIA(0): Initialized GPU GART. (II) NVIDIA(0): Setting mode "DFP:1680x1050_120+0+0" (II) Loading extension NV-GLX (II) NVIDIA(0): Initialized OpenGL Acceleration |
However, for 3D vision to work on many Quadro models, it is necessary to connect the IR emitter to the GPU using a mini-DIN-to-1/8-inch connector to get the synchronisation signal for the glasses. This cable was missing in my 3D Vision shipment (ordered in France), but the Nvidia support team quickly mailed me the necessary part.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
Actually, the questions listed below aren't so frequently-asked at all. As of January 2010, the program has been downloaded by about 15,000 people. I have had almost no problems/complaints since version 5.1. But here are the main points people have raised...
Hex uses the Stride program to calculate secondary structure element types. Stride needs at least 5 residues to work with. Hence it cannot assign secordary structures shorter than this, and Hex will therefore draw a red tube (which normally represents a loop) in such cases. Additionally, if a PDB file contains two chains with the same chain label, Stride will skip the second chain, and only the first chain will be drawn correctly in cartoon mode. This can happen quite often if one uses Hex to view previously calculated complexes, for example.
In general, the answer is no. At least not yet. Hex represents many years of work and it implements some ideas that have not been published yet. I would also like to keep open the possibility of potential future commercialisation, which might be difficult if the source is freely available. If your main interest in the source is just to poke around to see how it works, there are other OpenGL-based molecular graphics packages which do provide source that you could take a look at instead (e.g. VMD, MolMol). If you're more interested in the computational parts (spherical harmonics, Laguerre polynomials, etc.), these should be quite easy for any reasonably competent C or Fortran programmer to code up anyway. In terms of lines of code, this represents only a very small proportion of the program.
On the other hand, I can see that putting it all together to make a new docking program (or other application) is not trivial. I am considering making all the polar Fourier code available as a dynamic runtime library (DLL or .so). Please let me know if you are interested in this idea...
I'm not aware of any recent Intel Linux systems it doesn't run on. However, I can't guarantee that Hex will run on every Linux system, because a lot depends on where the run-time libraries are located (eg. /usr/lib/libc.so, etc.), although I've tried to minimise such dependencies.
There are really two main reasons why Hex might not work.
Assuming you have downloaded an appropriate executable, the problem is
probably due to incorrect file permissions or the executable search path.
If nothing happens when you type hex, its probably a problem with the
HEX_ROOT and PATH variables (check the README file). If these look OK,
change directory to the hex/exe directory and type ./hex6a.i586
to start the program without the aid of the
front-end startup script - it should give you a graphics
window with the Hex logo on a black background.
The 5 part of the filename
corresponds to version 5.0 - if you have a different version of the program,
the executables will have slightly different names.
Anyway, if that works, the
problem is definitely with HEX_ROOT or PATH - in which case, check that
HEX_ROOT is set to the directory you installed hex in (i.e. /your/stuff/hex
without anything after the hex bit): also check that the PATH variable
includes $HEX_ROOT/bin - i.e. /your/stuff/hex/bin in amongst the other
colon-separated bits of the PATH (sorry if this is all obvious,
but different people have different levels of knowledge). You should
be able to type which hex and get back /your/stuff/hex/bin
if the PATH is properly set up.
If running stand-alone ./hex5.i586 doesn't work, the next thing to try
is the ldd utility. This shows the dependency list of any shared
libraries that a program was linked against:
% ldd hex5d.i586 |
The output on my Linux system looks like this:
linux-gate.so.1 => (0x00b36000) libGLU.so.1 => /usr/X11R6/lib/libGLU.so.1 (0x028f3000) libGL.so.1 => /usr/lib/libGL.so.1 (0x00a7d000) libXext.so.6 => /usr/X11R6/lib/libXext.so.6 (0x00d6a000) libX11.so.6 => /usr/X11R6/lib/libX11.so.6 (0x00c94000) libpthread.so.0 => /lib/libpthread.so.0 (0x00d7b000) libstdc++.so.6 => /usr/lib/libstdc++.so.6 (0x008e7000) libm.so.6 => /lib/libm.so.6 (0x00c53000) libgcc_s.so.1 => /lib/libgcc_s.so.1 (0x008db000) libc.so.6 => /lib/libc.so.6 (0x00112000) libGLcore.so.1 => /usr/lib/libGLcore.so.1 (0x00d8d000) libnvidia-tls.so.1 => /usr/lib/tls/libnvidia-tls.so.1 (0x00b23000) libdl.so.2 => /lib/libdl.so.2 (0x00c79000) /lib/ld-linux.so.2 (0x00b05000) |
If there are any blanks in the ldd output or if the version numbers of the libraries in your /lib directory are hopelessly below the numbers here, then this is probably the problem. You would then need to try installing a version that was built for a more similar vintage of your operating system, or (better) you should update your entire operating system.
Otherwise, you're out of luck for the moment - but I'd appreciate it if
you could send me some details of your system (e.g. Linux vendor & release,
output from uname -a, etc.) and particularly the
exact message you get after typing ./hex5.i586 - I'll try to figure out
what needs to be done and if possible make up a new Linux release to fix it.
By default, Hex writes all its information and warning messages to
a dedicated text output window.
This window is is controlled by a separate subprocess
(so you will normally see two Hex processes running in
your system's process or task monitor tool), and Unix-style
sockets are used to communicate between the two processes. The error messages
referring to “bind” or “connect” indicate there was a problem creating
the message window subprocess.
This could happen if networking is disabled on your machine (unlikely),
if there is a problem with the machine's hostname (possible), or
on very slow machines (likely, due to time-outs).
Hex always assumes that the standard name localhost
(IP address 127.0.0.1) exists, and that networking is enabled to the
extent that ping localhost works.
On Unix/Linux machines, you can stop Hex
from trying to create a message window by typing
hex -nomessages,
or by setting the environment variable HEX_MESSAGES=no, in which case
Hex will print all its messages to the terminal window.
Windows XP does not use terminal windows, and Hex relies on the
message window process to show all text messages.
I compared the docking performance of Hex compiled for a 686 processor on my 686 system with a 585-compiled version, and there was hardly any difference. Docking performance depends almost entirely on the efficiency of a few floating point operations, and the larger 686 instruction set doesn't make floating point operations go any faster. Having said that, compiling with Intel's icc gives about a 10% speed-up over GNU's gcc-2.96, and the release versions of Hex are compiled with icc whenever possible (sorry, GNU).
The $HEX_ROOT/data directory contains several files (residue_types.dat, atom_bonds.dat atom_names.dat, atom_types.dat) which define the atom connectivity, charges, and names of non-standard residues. You will need to define new entries in these files to define your new residues. Probably the easiest approach is to identify the most similar existing amino acid and to copy/rename/edit the entries. You may need to calculate/guess some partial charges for the new atoms.
It is possible to further refine results from the docking correlation in Hex version 3. This “refinement” applies a soft molecular-mechanics energy evaluation and/or minimisation to the top 1000 orientations. However, if the amount of conformational change on binding is large, this refinement could eliminate what was a good solution. On the other hand, if the conformational change is small, then the calculation should lower the energy of a good orientation and significantly raise the energy of other orientations. I think its hard to assess how good the predicted orientations might be, and which of them might correspond to the actual solution. It is certainly hard to devise a scoring function which can reliably eliminate “false-positives” (Camacho & Vajda, Curr Op Struct Biol, 2002 12:36-40). Camacho & Vajda, and others, have proposed using a combination of algorithms, and to look for consensus orientations, but I have doubts. Personally, I think the surest approach is to use whatever experimental or bioinformatics evidence (e.g. correlated mutations) might be available to help suggest the location of the binding sites, and then to use a docking algorithm to create some feasible models which can subsequently be challenged by lab-based experiments.
Its not possible to do a pure electrostatic correlation in Hex. However, it should be possible to increase the weight of the electrostatic correlation in a shape + electrostatic calculation. For example, if you create a small macro file containing
factor_g 7000 |
and then run this macro using the Macros ... Run menu, all subsequent
docking calculations will have electrostatic energies scaled up by
a factor of 10. Thus electrostatics should dominate. Generally, I
would say that shape complementarity is more important that electrostatics,
but if you know electrostatics are important for a given system, then this
may be worth trying.
Its likely that your structures contain unrecognised residues, and/or unrecognised atoms. Hex assigns a default charge and radius to any unrecognised atoms, normally based on the first letter of each atom name. These defaults are usually reasonable, but if a structure has a large number of unrecognised hydrogens, Hex might assign 0.5e to each hydrogen to give a highly positively charge protein. A pair of such proteins will have a very unfavourable electrostatic interaction, and docking such a pair will give a total of zero low energy orientations. In order to avoid having zero-length arrays within the program, Hex forces at least one solution to be reported.
There is currently no licence fee for commercial users. But this does not mean the software is “free”. When you download or otherwise obtain a copy of Hex you are agreeing to be bound by the Hex Licence Agreement. You are also asked to obtain prior agreement with the author if the program is to be used for profit. This might seem to be unnecessarily complicated, but the intention is to make the software available and to publicise the algorithms used, without compromising the possibility of potential future commercialisation. These days, universities like to protect their assets, and I want to protect my assetc ;-).
In Hex, the reference (zero) energy point is for two proteins at infinite separation. i.e. zero interaction. Negative scores are favourable, positive unfavourable (i.e. the molecules interpenetrate, or have poor surface contacts). The electrostatic energy Hex calculates corresponds to the classical internal energy (U) of a system of point charges. The shape complementarity score, or “hydrophobic energy”, is scaled to comparable units (but forced to have more negative energies). This pseudo energy is also closer to U than G, but could easily be in error by 100's of KJ/mol compared to experimental measurements. I have compared calculated Hex energies of complexes with equilibrium constants (i.e. ignoring the difference between U and G due to entropy) for some antibody/antigen complexes and found no correlation. So, the calculated energies are pretty meaningless on an absolute scale. But I'm sure this is also true of most (all?) other macromolecular docking software.
The first few controls in the Docking Control panel define the search increments that Hex will use during a docking search. If you increase the number of steps (i.e. use more and smaller steps) then the rank of the “right” orientation is likely to be reduced because Hex will find more low-energy false-positive orientations during its search. This effect can largely be alleviated by ensuring you cluster the generated solutions using the Clustering Control panel. In general, its better to over-sample the search space and then cluster the solutions rather than risk missing good solutions by under-sampling in order to make the program run faster.
The default value of the Distance Range in the Docking Control panel may be too small for large protein-protein complexes. Hence, if you find that increasing the distance range gives a completely different set of docking orientations, its likely that you were seriously under-sampling the search space in the earlier run. In other words, if the distance range is too small, the program is not going to explore all possible surface-surface contacts as one molecule is rotated against the other.
Hex samples radial distances “symmetrically” from the starting orientation according to (Starting Distance) +/- (Distance Range)/2 in equal steps of +/-(Step Size). One way to work out what distance range to specify for a global search is to sketch each protein as a plane cross-section in such a way that the plane chosen for each protein includes its centroid, and the largest and smallest radial rays from the centroid to the surface. Then, set the initial inter-centroid distance (the starting distance) to the average of the sums of the smallest and largest radii. For example, if the smallest radii sum to 28A, and the largest radii sum to 46A, you should set the starting separation to (28+46)/2=37A, and the Distance Range to 2*(46-37)=18A. Using the default Step Size (0.75A), Hex will then explore 18/0.75=25 trial distances (i.e. 12 inward and 12 outward distance steps, plus 1 for the initial distance).
In practice, its often not possible to calculate the radii exactly, and hence set an optimal initial intermolecular separation. An alternative approach is to enable the Radial Envelope Filter, and to set a large Distance Range. When the radial filter is enabled, Hex will use low-resolution spherical harmonic surface approximations of each surface to select which intermolecular distances to evaluate at each angular pose of each molecule. However, because these surface representations are inexact (they assume the proteins are globular, or star-like), there is a possibility that some orientations that should be evaluated might be skipped.
Hex should always place the ligand near the receptor in tightly fitting orientations. You should get a ranked list of such orientations at the end of each run, and you should be able to scroll through these interactively using the Page/Down and Page/Up (and Home and End) keys on the keyboard. Whether any of these orientientations actually represent the right answer, is of course, a completely separate matter!! In any case, if you are not seeing this, it is probably because you need to place the two molecules quite close together before starting the calculation. Hex searches over the range of intermolecular separations and angular orientations as specified in the Docking Control panel. If the molecules are initially too far apart, Hex will not search over (relatively) small separations unless you specify a large distance range in the docking search. But this will add significantly to the execution time. If you know the location of the ligand binding site, you should manually move the ligand to be close to the binding site (this can be done in Edit Molecule mode), and then restrict the receptor rotation angle range to a small value, say 45 degrees. If you know (or have a hunch about) the binding mode of the ligand, you can also use Edit Molecule mode to orient the binding surface of the ligand towards the receptor binding site, and then also restrict the ligand rotation angle similarly. When pre-positioning the molecules, it may help to enable the inter-molecular axis display (the double-headed arrow on the right-hand border). This shows the current z-axis as a white line, about which the docking is performed. The end-points of this line are the molecular origins (which can also be changed using Edit Origin mode). Please also look at the answer given for the previous FAQ.
Etot is the total calculated interaction energy of the system. Hex doesn't calculate internal energies, and so doesn't report separate individual molecular energies.
If you select File ... Save ... Range from the File menu,
you can write
out each prediction of the complex to a separate PDB file - these can
then be read back into Hex in subsequent sessions.
However, these are then treated as a "single" molecule in Hex,
rather than the seprate receptor + ligand that you started with.
Alternatively, you can select a docking prediction, and then use
File ... Save ... Receptor and File ... Save ... Ligand
to save the separate
components of the current orientation. These can then be
read back together from the Unix command line:
hex receptor.pdb ligand.pdb |
Unfortunately, its not possible to reload together all the predictions from a previous session. You would need to rerun the calculation from the beginning to get that behaviour.
No, Hex can exploit only the processors ot it can see on the motherboard. Since the basic calculation now takes only a matter of seconds, there is little point in using multiple nodes for a single pair-wise docking calculation. If you wish to do multiple pair-wise docking, I would recommend using Unix scripts to spread the calculation over multiple nodes on a cluster and to collect the results.
Quite a few journal articles cite Hex. However, they generally do not describe in detail how it was used, and so would probably not help someone to get started. If you do wish to look at articles that use Hex, you would need to use a bibliography service to search for papers that cite Ritchie & Kemp.
This bug should have been fixed in Hex version 4.5. If you cannot upgrade to that version (or later), you probably need to edit the ligand PDB file to change "HETATM" to "ATOM " (6 characters). Prior to version 4.5, Hex would not display a molecule that contains only HETATM data.
The "-1" means nothing was calculated. You will always see this value unless you enable Backbone Bumps in the Postprocessing selector in the Docking Control panel. In that case, each orientation will be checked for steric clashes between backbone atoms (the column will show the total count of bumps). You can then use the Clustering control panel to put solutions with more than a given number of bumps to the end of the list, if you wish. Requesting molecular mechanics (MM) energies also causes backbone bumps checking to be turned on. Similarly, "Hbonds = -1" means no hydrogen bonds were calculated. You need to enable the Show H-bonds toggle in the Molecule Control panel.
| [ < ] | [ > ] | [ << ] | [ Up ] | [ >> ] | [Top] | [Contents] | [Index] | [ ? ] |
| Jump to: | 5
A B C D E F G H I K L M N O P R S T V W Z |
|---|
| Jump to: | 5
A B C D E F G H I K L M N O P R S T V W Z |
|---|
| [Top] | [Contents] | [Index] | [ ? ] |
| [Top] | [Contents] | [Index] | [ ? ] |
This document was generated by Dave Ritchie on November, 28 2010 using texi2html 1.78.
The buttons in the navigation panels have the following meaning:
| Button | Name | Go to | From 1.2.3 go to |
|---|---|---|---|
| [ < ] | Back | Previous section in reading order | 1.2.2 |
| [ > ] | Forward | Next section in reading order | 1.2.4 |
| [ << ] | FastBack | Beginning of this chapter or previous chapter | 1 |
| [ Up ] | Up | Up section | 1.2 |
| [ >> ] | FastForward | Next chapter | 2 |
| [Top] | Top | Cover (top) of document | |
| [Contents] | Contents | Table of contents | |
| [Index] | Index | Index | |
| [ ? ] | About | About (help) |
where the Example assumes that the current position is at Subsubsection One-Two-Three of a document of the following structure:
This document was generated by Dave Ritchie on November, 28 2010 using texi2html 1.78.